Organic catalysis is a major topic of Chapter 5 of my book. The use of iminium ions as a catalyst and to provide stereoselection, pioneered by MacMillan,1 was not discussed in the book.
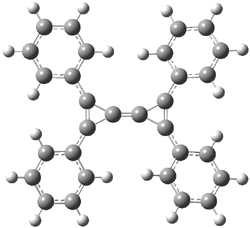
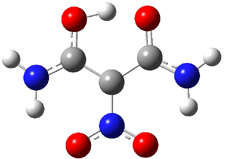
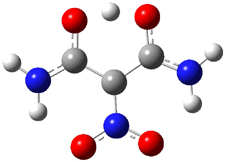
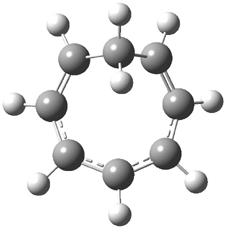
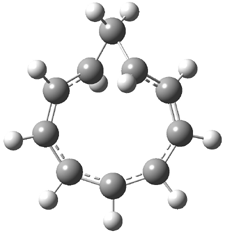
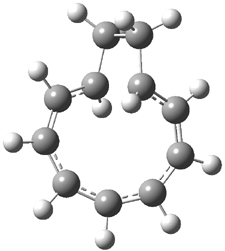
Macmillan had proposed that the iminium 2 formed of imidazolinone 1 and (E)-3-phenylprop-2-enal has conformation A. This conformation blocks access to one face of the alkene and directs, for example, dienophiles to the opposite face. Houk found that conformer B is lower in energy at B3LYP/6-31G(d).2
|
|
|
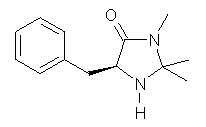
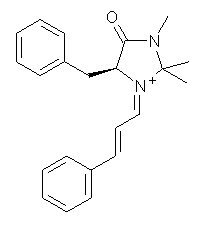
Now Tomkinson3 has produced a study that convincingly shows that 2 exists as conformer B. An x-ray structure shows this conformation in the solid state. Proton NMR shows that the methyl group signals are interpretable only as coming from B. Finally, SCS-MP2/aug-cc-pVTZ//BHandH/6-31+G(d,p) (see Figure 1) computations show that B is 1.2 kcal mol-1 more stable than A in the gas phase, and PCM computations indicate that this gap is reduced by less then 0.5 kcal mol-1 in methanol or acetonitrile.
Conformation B provides little steric hindrance at the β-carbon of the iminium ion, explaining its poor stereoselectivity in conjugate additions.
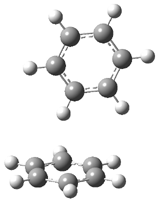
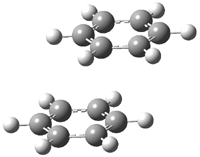
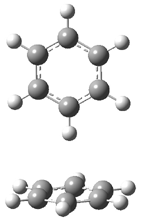
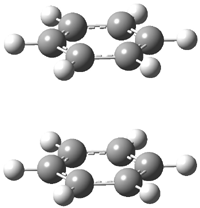
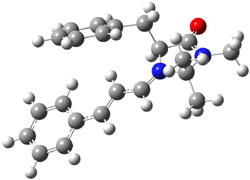 A |
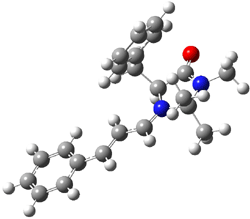 B |
Figure 1. BHandH/6-31+G(d,p) optimized structures of conformers A and B of 2.
References
(1) Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C., "New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels-Alder Reaction," J. Am. Chem. Soc., 2000, 122, 4243-4244, DOI: 10.1021/ja000092s.
(2) Gordillo, R.; Houk, K. N., "Origins of Stereoselectivity in Diels-Alder Cycloadditions Catalyzed by Chiral Imidazolidinones," J. Am. Chem. Soc., 2006, 128, 3543-3553, DOI: 10.1021/ja0525859.
(3) Brazier, J. B.; Evans, G.; Gibbs, T. J. K.; Coles, S. J.; Hursthouse, M. B.; Platts,
J. A.; Tomkinson, N. C. O., "Solution Phase, Solid State, and Theoretical Investigations on the MacMillan Imidazolidinone," Org. Lett., 2009, 11, 133-136, DOI: 10.1021/ol802512y.
InChIs
1: InChI=1/C13H18N2O/c1-13(2)14-11(12(16)15(13)3)9-10-7-5-4-6-8-10/h4-8,11,14H,9H2,1-3H3/t11-/m0/s1
InChIKey=UACYWOJLWBDSHG-NSHDSACABQ
2: InChI=1/C22H25N2O/c1-22(2)23(3)21(25)20(17-19-13-8-5-9-14-19)24(22)16-10-15-18-11-6-4-7-12-18/h4-16,20H,17H2,1-3H3/q+1/b15-10+,24-16+/t20-/m0/s1
InChIKey=ZPEHVNACGWTABV-BYFMJTDEBT