I have written many posts on the use of computed NMR shifts as a tool for determining molecular structure, especially stereochemistry. All of these methods rely upon computing a bunch of alternative structures and then identifying the one whose chemical shifts (1H and/or 13C) match up best with experiment. Many people have been interested in the first part of this process – the “computing a bunch of alternative structures” – testing the QM method, the basis set, the selection of conformation(s), and the method for computing chemical shifts. The subject of this post is the notion of “matching up best” and comes from of a recent article by Jonathan Goodman.1
So in the typical procedure for deciding which structure (of many) best accounts for the experimental NMR spectra, the computed NMR shifts (and perhaps coupling constants) are compared to the experimental data. This comparison is done often by simply examining the correlation coefficient r between the experimental and calculated shifts. Some have used the mean absolute error between the computed and experimental shifts. Others have employed a corrected mean absolute error where scaled chemical shifts are first obtained from the plot of the calculated vs. experimental shifts, and then finding the average of the differences between these scaled shifts and the experimental ones.
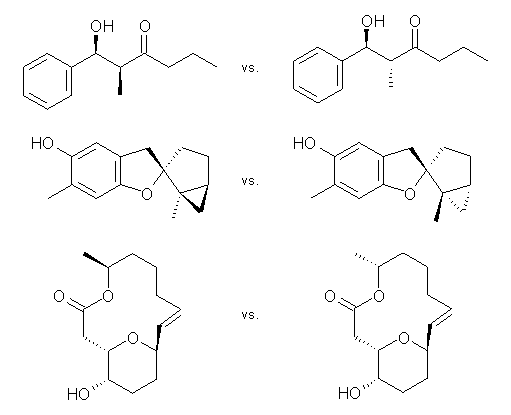
Goodman suggests that oftentimes what is of interest is not really the chemical shifts of a compound but rather identifying the structure of diastereomers, and then it’s really the differences in the chemical shifts of pairs of diastereomers that are really critical in identifying which one is which. Using Goodman’s notation, suppose you have experimental NMR data on diastereomers A and B and the computed NMR shifts for structures a and b. The key is deciding does A correlate with a or b and the same for B. Goodman proposes three variants on how to compare the chemical shift differences, but I’ll show just the first, which he calls CP1. Define Δexpi as the differences in the experimental chemical shifts of the two diastereomers for nucleus i: Δexp = δAi – δBi and a similar definition for the differences in the computed shifts: Δcalc = δai – δbi. CP1 is then defined as Σ (Δexp/Δcalc)/Σ (Δexp)2 where each sum is over the nuclei i. Goodman shows in a number of examples (some are shown below) that CP1 and its variants provides an excellent measure of when a computed structure’s chemical shifts agree with the experimental values, along with a means for noting the confidence in that assignment. These CP measures provide significantly better measures of agreement that the ones previous utilized, providing a real confidence level in assessing the quality of the prediction. I strongly urge all who are interested in the use of computed NMR in determining molecular structures to read this paper and consider adopting this approach.
References
(1) Smith, S. G.; Goodman, J. M., "Assigning the Stereochemistry of Pairs of Diastereoisomers Using GIAO NMR Shift Calculation," J. Org. Chem. 2009, 74, 4597-4607, DOI: 10.1021/jo900408d