Yang, Ganz, Chen, Wang, and Schleyer have published a very interesting and comprehensive review of planar hypercoordinate compounds, with a particular emphasis on planar tetracoordinate carbon compounds.1 A good deal of this review covers computational results.
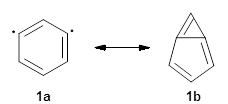
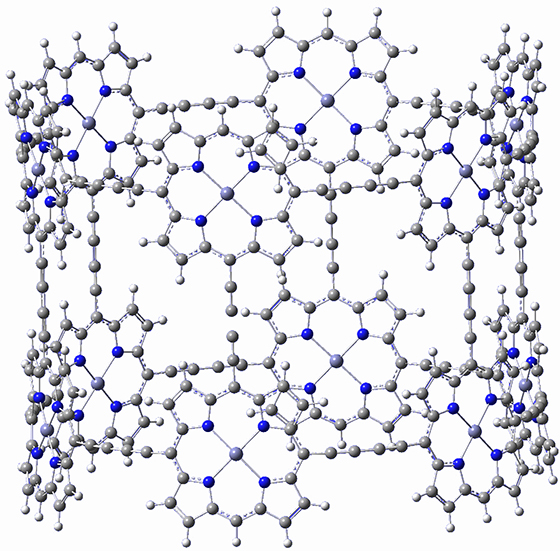
There are two major motifs for constructing planar tetracoordinate carbon compounds. The first involves some structural constraints that hold (or force) the carbon into planarity. A fascinating example is 1 computed by Rasmussen and Radom in 1999.2 This molecule taxed their computational resources, and as was probably quite typical for that time, there is no supplementary materials. But since this compound has high symmetry (D2h) I reoptimized its structure at ω-B97X-D/6-311+G(d) and computed its frequencies in just a few hours. This structure is shown in Figure 1. However, it should be noted that at this computational level, 1 possesses a single imaginary frequency corresponding to breaking the planarity of the central carbon atom. Rasmussen and Radom computed the structure of 1 at MP2/6-31G(d) with numerical frequencies all being positive. They also note that the B3LYP/6-311+G(3df,2p) structure also has a single imaginary frequency.
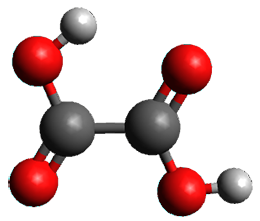
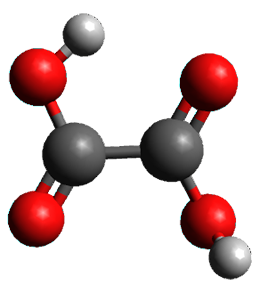
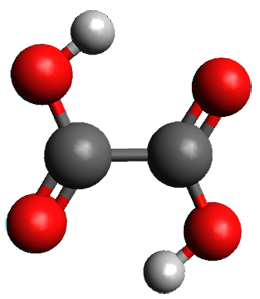
A second approach toward planar tetracoordinate carbon compounds is electronic: having π-acceptor ligands to stabilize the p-lone pair on carbon and σ-donating ligands to help supply sufficient electrons to cover the four bonds. Perhaps the premier simple example of this is the dication 2¸ whose ω-B97X-D/6-311+G(d,p) structure is also shown in Figure 1.
The review covers heteroatom planar hypercoordinate species as well. It also provides brief coverage of some synthesized examples.
|
|
|
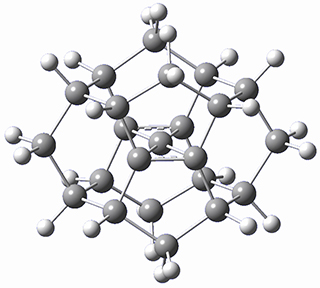
1 |
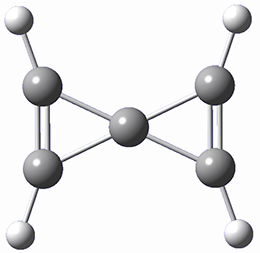
2 |
Figure 1. Optimized structures of 1 and 2.
References
(1) Yang, L.-M.; Ganz, E.; Chen, Z.; Wang, Z.-X.; Schleyer, P. v. R. "Four Decades of the Chemistry of Planar Hypercoordinate Compounds," Angew. Chem. Int. Ed. 2015, 54, 9468-9501, DOI: 10.1002/anie.201410407.
(2) Rasmussen, D. R.; Radom, L. "Planar-Tetracoordinate Carbon in a Neutral Saturated Hydrocarbon: Theoretical Design and Characterization," Angew. Chem. Int. Ed. 1999, 38, 2875-2878, DOI: 10.1002/(SICI)1521-3773(19991004)38:19<2875::AID-ANIE2875>3.0.CO;2-D.
InChIs
1: InChI=1S/C23H24/c1-7-11-3-15-9-2-10-17-5-13-8(1)14-6-18(10)22-16(9)4-12(7)20(14,22)23(22)19(11,13)21(15,17)23/h7-18H,1-6H2
InChIKey=LMDPKFRIIOUORN-UHFFFAOYSA-N
2: InChI=1S/C5H4/c1-2-5(1)3-4-5/h1-4H/q+2
InChIKey=UGGTXIMRHSZRSQ-UHFFFAOYSA-N