Well, here’s my vote for paper of the year (at least so far!). It is work from Barry Carpenter’s lab1 and pertains to many topics discussed in my book, including pericyclic and psuedopericylic reactions, non-statistical dynamics, and the use of high-level computations to help understand confusing experimental results. The paper is in an interesting read – and not just for the great science. It is told as a story, recounting the experiments and interpretation as they took place in chronological order with a surprising and critical contribution made from a referee!
The story begins with Carpenter’s continuing interest in unusual dynamic effects and the supposition that non-statistical dynamics might be observed in the rearrangements of carbenes. So, they took on the Wolff rearrangement, specifically the rearrangement of 3 into 4. Using labeled starting material 1, one should observe equal amounts of 4a and 4b if normal statistical dynamics is occurring (Scheme 1).
Scheme 1.

In fact, the ratio of products is not unity, but rather 4a:4b = 1:4.5. But the excess of 4b could be the result of another parallel rearrangement, 2 to 5 to 4b (Scheme 2).
Scheme 2.
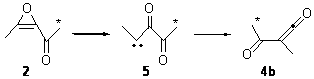
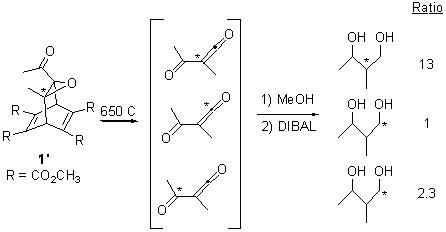
To try to distinguish whether 5 is intervening, they carried out the photolysis of a different labeled version of 1 (namely 1’). The product distribution of the products is shown in Sheme 3. It appears that the reaction through 5 dominates, but the ratio of products that come from 3 still shows non-statistical behavior.
Scheme 3.
CCSD(T) computation suggested that 5 is higher in energy than 3, and this does not help understand the experiments. At this point, Carpenter decided to write up the work as a communication, with the main point that non-statistical dynamics were occurring.
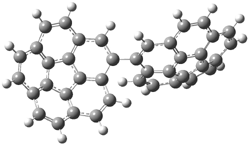
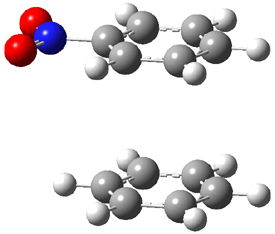
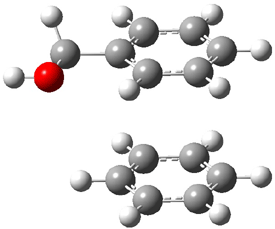
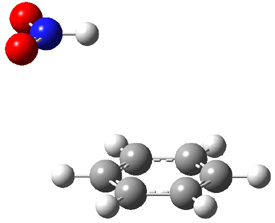
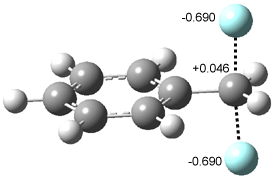
Now here an unusual event took place that offers up hope that the peer-review system still works! A referee, later identified as Dan Singleton, offered an alternative mechanism for the production of 5. Shown in Scheme 4 is the novel pseudopericylic reaction that leads from 1 directly to 5. In fact, the transition state for this pseudopericyclic reaction is 19.0 kcal mol-1 lower in energy than the transition state for the retro-Diels-Alder reaction of Scheme 1 (computed at MPWB1K/ 6-31+G(d,p), and this pseudopericyclic TS is shown in Figure 1).
Scheme 4.
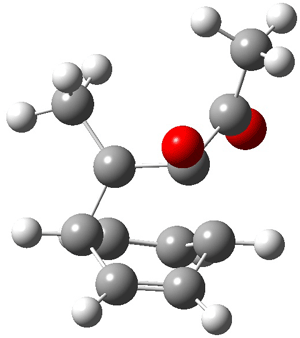 |
Figure 1. MPWB1K/6-31+G(d,p) optimized geometry of the transition state for the pseudopericyclic reaction shown in Scheme 4.1
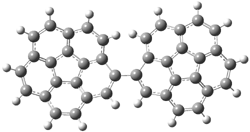
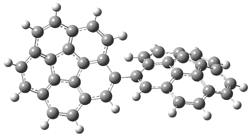
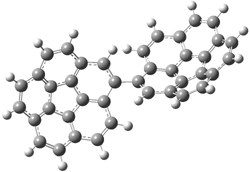
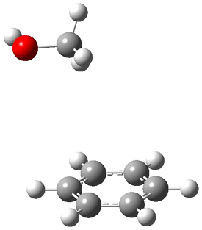
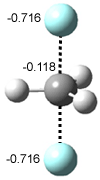
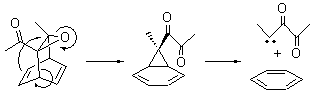
The revised mechanism was then modified to include the additional complication of the formation of 6, and is shown in Scheme 5, along with their relative CCSD(T) energies. The CCSD(T)/cc-pVTZ//CCSD/cc-pVTZ optimized geometries of the critical points of Scheme 5 are drawn in Figure 2.
Scheme 5.

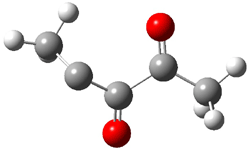 5 |
|
|
TS 5 → 3 |
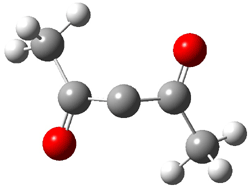 3 |
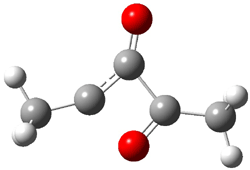 TS 5 → 6 |
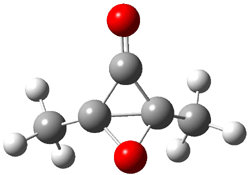 6 |
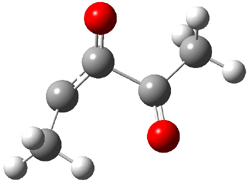 TS 5 → 4 |
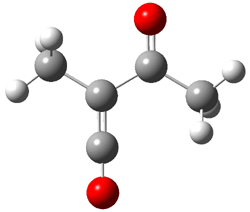 4 |
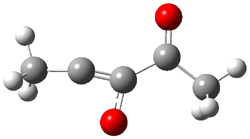
Figure 2. CCSD/cc-pVTZ optimized geometries.1
Any non-statistical effect would occur in the transition from 5 to 3. A direct dynamics trajectory analysis was performed starting in the neighborhood of this TS using three different functionals to generate the potential energy surface. Though only 100 trajectories were computed, the results with all three functionals are similar. About 2/3rds of these trajectories led to 3 followed by the shift of the C5 methyl group. Another 15% led to 3 and then the C1 methyl shifted. This MD simulation supports the non-statistical Wolff rearrangement, with a clear preference for the C5 shift, consistent with experiment. A larger MD study is underway and will hopefully shed additional insight onto this fascinating reaction.
References
(1) Litovitz, A. E.; Keresztes, I.; Carpenter, B. K., "Evidence for Nonstatistical Dynamics in the Wolff Rearrangement of a Carbene," J. Am. Chem. Soc., 2008, 130, 12085-12094, DOI: 10.1021/ja803230a.
InChIs
3: InChI=1/C5H6O2/c1-4(6)3-5(2)7/h1-2H3
InChIKey = IGYQBMPIQGLNRU-UHFFFAOYAO
4: InChI=1/C5H6O2/c1-4(3-6)5(2)7/h1-2H3
InChIKey = ABVJXABNYINQLN-UHFFFAOYAA
5: InChI=1/C5H6O2/c1-3-5(7)4(2)6/h1-2H3
InChIKey = FJJXVDYICOYKRN-UHFFFAOYAD
6: InChI=1/C5H6O2/c1-4-3(6)5(4,2)7-4/h1-2H3
InChIKey = CAMBQRQJZBTNNS-UHFFFAOYAB