Following on previous work (see these posts on ladderane and hexaphenylethane), Schreiner, Grimme and co-workers have examined the structure of the all-meta tri(di-t-butylphenyl)methane dimer 12.1 In the study of hexaphenylethane,2 Schreiner and Grimme note that t-butyl groups stabilize highly congested structures through dispersion, identifying them as “dispersion energy donors”.3 The idea here is that the dimer of 1 will be stabilized by these many t-butyl groups. In fact, the neutron diffraction study of the crystal structure of 12 shows an extremely close approach of the two methane hydrogens of only 1.566 Å, the record holder for the closest approach of two formally non-bonding hydrogen atoms.
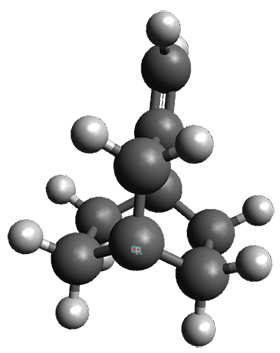
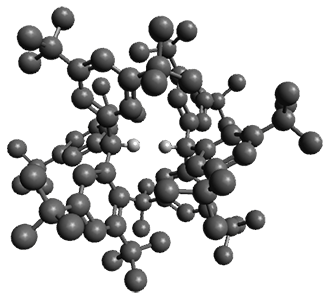
To understand the nature of this dimeric structure, they employed a variety of computational techniques. (Shown in Figure 1 is the B3LYPD3ATM(BJ)/def2-TZVPP optimized geometry of 12.) The HSE-3c (a DFT composite method) optimized crystal structure predicts the H…H distance is 1.555 Å. The computed gas phase structure lengthens the distance to 1.634 Å, indicating a small, but essential, role for packing forces. Energy decomposition analysis of 12 at B3LYP-D3ATM(BJ)/def2-TZVPP indicates a dominant role for dispersion in holding the dimer together. While 12 is bound by about 8 kcal mol-1, the analogue of 12 lacking all of the t-butyl groups (the dimer of triphenylmethane 22) is unbound by over 8 kcal mol-1. Topological electron density analysis does show a bond critical point between the two formally unbound hydrogen atoms, and the noncovalent interaction plot shows an attractive region between these two atoms.
|
Figure 1.
References
1) Rösel, S.; Quanz, H.; Logemann, C.; Becker, J.; Mossou, E.; Cañadillas-Delgado, L.; Caldeweyher, E.; Grimme, S.; Schreiner, P. R., "London Dispersion Enables the Shortest Intermolecular Hydrocarbon H···H Contact." J. Am. Chem. Soc. 2017, 139, 7428–7431, DOI: 10.1021/jacs.7b01879.
2) Grimme, S.; Schreiner, P. R., "Steric Crowding Can Stabilize a Labile Molecule: Solving the Hexaphenylethane Riddle." Angew. Chem. Int. Ed. 2011, 50 (52), 12639-12642, DOI: 10.1002/anie.201103615.
3) Grimme, S.; Huenerbein, R.; Ehrlich, S., "On the Importance of the Dispersion Energy for the Thermodynamic Stability of Molecules." ChemPhysChem 2011, 12 (7), 1258-1261, DOI: 10.1002/cphc.201100127.
InChIs
1: InChI=1S/C43H64/c1-38(2,3)31-19-28(20-32(25-31)39(4,5)6)37(29-21-33(40(7,8)9)26-34(22-29)41(10,11)12)30-23-35(42(13,14)15)27-36(24-30)43(16,17)18/h19-27,37H,1-18H3
InChIKey=VFNQDWKFTWSJAU-UHFFFAOYSA-N