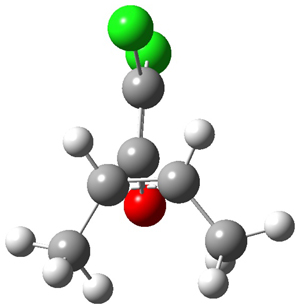
I seem to be recently flooded with papers dealing with tunneling in organic systems. Well, here’s one more! Kozuch, Borden, Schreiner and co-workers seek out systems whereby isotopic substitution might lead to reaction selectivity.1 Their base system is cyclopropylmethylcarbene 1, which can undergo three different reactions: (a) the ring can expand to give 1-methylcyclobut-1-ene 2, (b) a hydrogen can shift from the terminal methyl group to give vinylcyclopropane 3, or (c) the methane hydrogen can shift to produce ethylidenecyclopropane 4. This last option can be neglected since its barrier (20.5 kcal mol-1) is so much higher than for the other two, 7.5 kcal mol-1 for the ring expansion and 12.1 kcal mol-1 for the [1,2]H-shift converting 1 → 3.
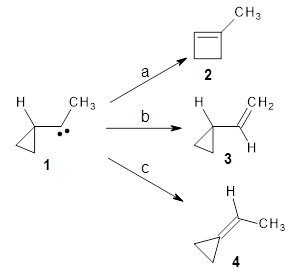
At high temperature, the ring expansion to 2 will dominate, but at low temperature the hydrogen shift to 3 might dominate by tunneling through the barrier due to the low mass and short distances involved. The reaction rates were computed using B3LYP/6-31G(d,p) and small-curvature tunneling. At low temperature, the rate for the hydrogen shift is 10 orders of magnitude faster than the ring expansion. Thinking that deuterium substitution of the terminal methyl group might slow down the rate of the [1,2]-shift, they computed the rates for the reactions of 1-d3, and in fact the rate of this shift does reduce by 104 but it is still much faster than the rate for ring expansion. What is needed is a system where the rate for ring expansion is slower than the rate for hydrogen migration but faster than the rate of deuterium migration.
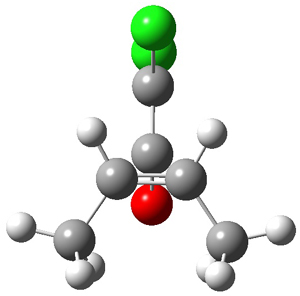
They examine a number of different substituents that may help to lower the barrier for the ring expansion. The methoxy derivative 5 turns out to suit the bill perfectly. The methoxy group reduces the barrier for ring expansion from 7.5 kcal mol-1 with 1 to 2.5 kcal mol-1 with 5. With hydrogenated 5, the [1,2]H-shift is 103 times faster than ring expansion, but with deuterated 5, ring expansion is twice as fast as the deuterium migration.
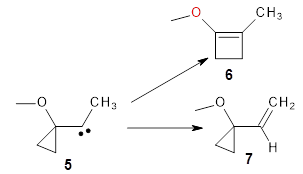
The authors call this isotope controlled selectivity (ICS), and this is the first example of this type of control.
References
1. Nandi, A.; Gerbig, D.; Schreiner, P. R.; Borden, W. T.; Kozuch, S., Isotope-Controlled Selectivity by Quantum Tunneling: Hydrogen Migration versus Ring Expansion in Cyclopropylmethylcarbenes. J. Am. Chem. Soc. 2017, 139, 9097-9099, DOI: 10.1021/jacs.7b04593.
InChIs
1: InChI=1S/C5H8/c1-2-5-3-4-5/h5H,3-4H2,1H3
InChIKey=KJIJNBZLGHBOTI-UHFFFAOYSA-N
2: InChIInChIKey=AVPHQXWAMGTQPF-UHFFFAOYSA-N
3: InChI=1S/C5H8/c1-2-5-3-4-5/h2,5H,1,3-4H2
InChIKey=YIWFBNMYFYINAD-UHFFFAOYSA-N
4: InChI=1S/C5H8/c1-2-5-3-4-5/h2H,3-4H2,1H3
InChIKey=ZIFNDRXSSPCNID-UHFFFAOYSA-N
5: InChI=1S/C6H10O/c1-3-6(7-2)4-5-6/h4-5H2,1-2H3
InChIKey=YMBSTCICUAORNN-UHFFFAOYSA-N
6: InChIInChIKey=QBLNAZHAVPMLHB-UHFFFAOYSA-N
7: InChIInChIKey=FHYLDABSPVPDTJ-UHFFFAOYSA-N