A recent paper by Papov, Shao, Bagdasarian, Benton, Zou, Yang, Houk, and Nelson uncovers a vinyl cation insertion reaction that once again involves dynamic effects.1
They find that vinyl triflates and cyclic vinyl triflates will react with [Ph3C]+[HCB11Cl11]– and triethylsilane to generate vinyl cations that can then be trapped through a C-H insertion reaction. For example, cyclohexenyl triflate 1 reacts in a cyclohexane solvent to give the insertion product 2.
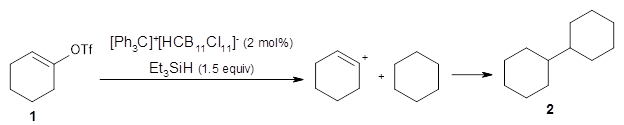
The reactions of isomers 3 and 4 give different ratios of the two products 5 and 6. In both cases, the cyclohexyl is trapped predominantly at the site of the triflate substituent. This means that the mechanism cannot involve a cyclohexene intermediate, since then the two ratios should be identical.
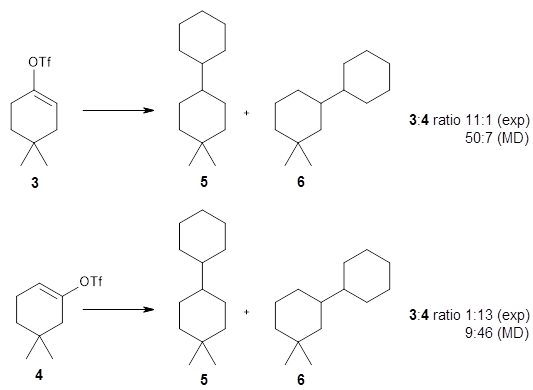
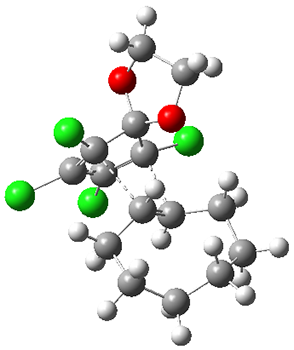
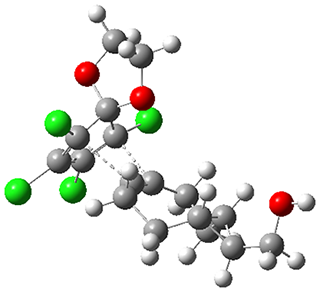
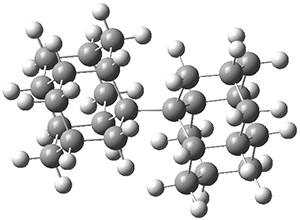
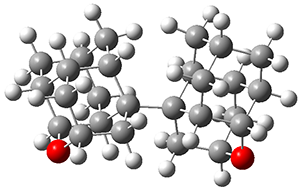
They performed molecular dynamic trajectory analysis at the M062X/6-311+G(d,p) level, starting with the two transition states leading from 3 (TS3) and 4 (TS4), the only transition states located for the insertion reaction. The structures of these TSs are shown in Figure 1.
|
|
|
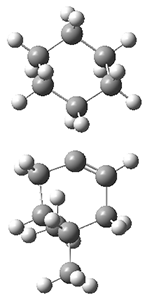
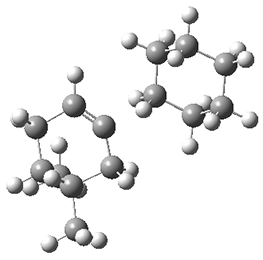
Figure 1. M062X/6-311+G(d,p) optimized geometries of TS3 and TS4.
The trajectories end up in two product basins associated with 5 and 6 starting with either TS3 or TS4. Thus, these transition states are ambimodal, and typical of reactions where dynamic effects dominate. For the reaction of 3, the majority of the trajectories starting at TS3 end up as 5, consistent with the experiments. Similarly, for the trajectories that start at TS4, the majority end up as 6, consistent with experiments.
Once again, we see that relatively simple organic reactions do not follow simple reaction mechanisms, that a single transition state leads to two different products and the product distributions are dependent on reaction dynamics. This may not be too surprising for the vinyl cation insertions given the many examples provide by the Tantillo group of cation rearrangements that are controlled by reaction dynamics (see for examples, this post and this post).
References
1. Popov, S.; Shao, B.; Bagdasarian, A. L.; Benton, T. R.; Zou, L.; Yang, Z.; Houk, K. N.; Nelson, H. M., "Teaching an old carbocation new tricks: Intermolecular C–H insertion reactions of vinyl cations." Science 2018, 361, 381-387, DOI: 10.1126/science.aat5440.
InChIs
1: InChI=1S/C7H10F3O3S/c8-7(9,10)14(11,12,13)6-4-2-1-3-5-6/h4H,1-3,5H2,(H,11,12,13)
InChIKey=CMPVYBNXADJVOM-UHFFFAOYSA-N
2: InChIInChIKey=WVIIMZNLDWSIRH-UHFFFAOYSA-N
3: InChI=1S/C9H14F3O3S/c1-8(2)5-3-7(4-6-8)16(13,14,15)9(10,11)12/h3H,4-6H2,1-2H3,(H,13,14,15)
InChIKey=XDWBLRRAHKBZJR-UHFFFAOYSA-N
4: InChI=1S/C9H14F3O3S/c1-8(2)5-3-4-7(6-8)16(13,14,15)9(10,11)12/h4H,3,5-6H2,1-2H3,(H,13,14,15)
InChIKey=YHVCPSRICQJFDT-UHFFFAOYSA-N
5: InChI=1S/C14H26/c1-14(2)10-8-13(9-11-14)12-6-4-3-5-7-12/h12-13H,3-11H2,1-2H3
InChIKey=BZQBWUOXOYWYJC-UHFFFAOYSA-N
6: InChI=1S/C14H26/c1-14(2)10-6-9-13(11-14)12-7-4-3-5-8-12/h12-13H,3-11H2,1-2H3
InChIKey=AENMAOBTECURBO-UHFFFAOYSA-N