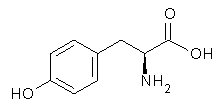
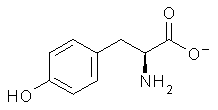
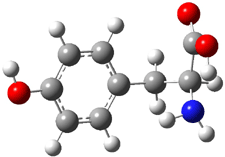
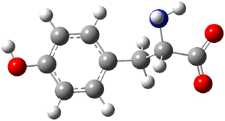
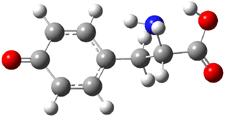
Molecular structures can differ depending on phase, particularly between the gas and solution phase. Kass has looked at the protonation of 4-aminobenzoic acid. In water, the amino is its most basic site, but what is it in the gas phase? The computed relative energies of the protonation sites are listed in Table 1. If one corrects the B3LYP values for their errors in predicting the proton affinity of aniline and benzoic acid, the carbonyl oxygen is predicted to be the most basic site by 5.0 kcal mol-1, in nice accord with the G3 prediction of 4.1 kcal mol-1. Clearly, the structure depends on the medium.
Table 1. Computed relative proton affinities (kcal mol-1) of 4-aminobenzoic acid.
| protonation site |
Erel B3LYP |
Erel G3 |
| C=O | 0.0 | 0.0 |
| NH2 | 7.9 | 4.1 |
| OH | 12.2 | 9.8 |
Electrospray of 4-aminobenzoic acid from 3:1 methanol/water and 1:1 acetonitrile/water solutions gave different CID spectra. H/D exchange confirmed that electrospray from the emthanol/water solution gave the oxygen protonated species while that from the acetonitrile/water solution gave the ammonium species.
References
(1) Tian, Z.; Kass, S. R., “Gas-Phase versus Liquid-Phase Structures by Electrospray Ionization Mass Spectrometry,” Angew. Chem. Int. Ed., 2009, 48, 1321-1323, DOI: 10.1002/anie.200805392.
InChIs
4-aminobenzoic acid: InChI=1/C7H7NO2/c8-6-3-1-5(2-4-6)7(9)10/h1-4H,8H2,(H,9,10)/f/h9H
InChIKey=ALYNCZNDIQEVRV-BGGKNDAXCD