While the [2-3]-sigmatropic rearrangement is well known and understood as allowed under the Woodward-Hoffmann rules, [1,2]-sigmatropic are much more rare, perhaps because they are forbidden by the same orbital symmetry arguments. It is perhaps surprising that these two rearrangements may sometimes be found in competition. Singleton has applied many of his tried-and-true techniques, namely, careful normal abundance kinetic isotope effect (KIE) analysis and molecular dynamics computations, to this problem.1
Reaction 1 takes place exclusively through a [2,3]-rearrangement; the principle evidence is the lack of any crossover reaction. However, the slightly more substituted analogue shown in Reaction 2 gives rise to two products: that obtained from a [2,3]-rearrangement 6 and that obtained from a [1,2]-rearrangement 7.
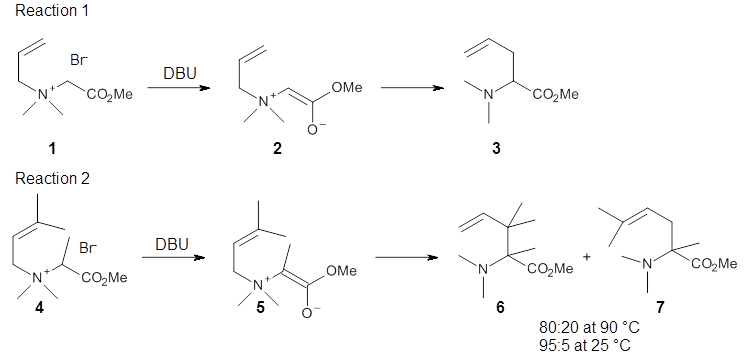
The KIE for the rearrangement of 2 is large for the carbon breaking the bond with nitrogen, while it is small at the carbons that are forming the new bond. This becomes a metric for judging the transition state obtained with computations. With the computed TS and canonical variational transition state theory (VTST) including small curvature tunneling, the KIE can be computed from a computed structures and frequencies. This imposes a range of reasonable distances for the forming C-C bond of 2.6-2.9 Å – much longer that a typical distance in the TS of similar pericyclic reactions.
Crossover experiments for Reaction 2 are understood in terms of a reaction model whereby some fraction of the reactants undergo a concerted rearrangement to form 6, and 7 is formed by first breaking the C-N bond, forming two radicals, that either recombine right away or form isolated radicals that then collapse to product.
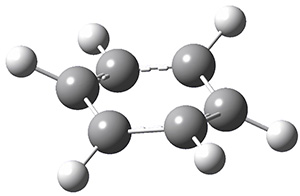
The interesting twist here is that one would expect two different transition states, one for the concerted process 8 and one to cleave the bond 9. Both do exist and are shown in Figure 1. However, VTST predicts that the concerted process should be 25-50 times faster than cleavage, and that does not match up with experiments. Amazingly, molecular dynamics trajectories started from the concerted TS 8 leads to cleavage about 20% of the time using UMO6-2X with a variety of basis sets. Thus, as Singleton has noted many times before, a single TS is crossed that leads to two different products! An argument based on entropy helps explain why the second (cleavage) pathway is viable.
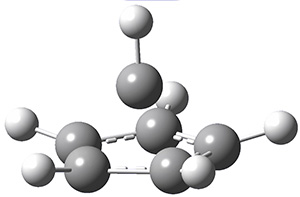
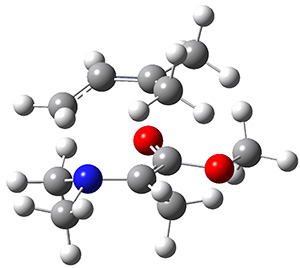 8 |
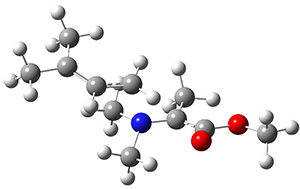 9 |
Figure 1. UMO6-2x/6-31G* optimized structures of TS 8 and 9.
References
(1) Biswas, B.; Collins, S. C.; Singleton, D. A. "Dynamics and a Unified Understanding of Competitive [2,3]- and [1,2]-Sigmatropic Rearrangements Based on a Study of Ammonium Ylides," J. Am. Chem. Soc. 2014, 136, 3740-3743, DOI: 10.1021/ja4128289.