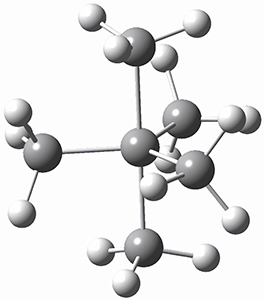
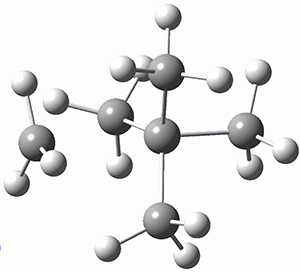
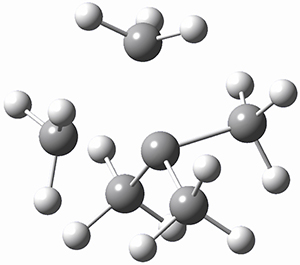
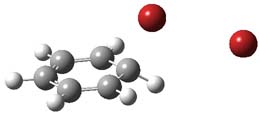
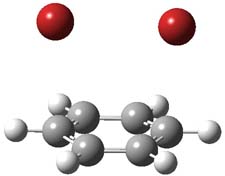
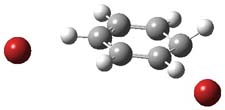
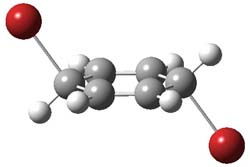
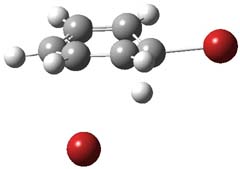
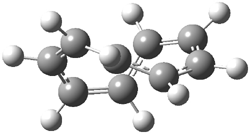
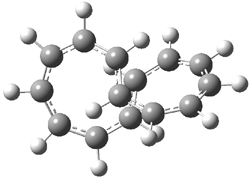
I want to update my discussion of m-benzyne, which I present in my book in Chapter 5.5.3. The interesting question concerning m-benzyne concerns its structure: is it a single ring structure 1a or a bicyclic structure 1b? Single configuration methods including closed-shell DFT methods predict the bicylic structure, but multi-configuration methods and unrestricted DFT predict it to be 1a. Experiments support the single ring structure 1a.
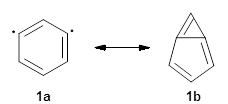
The key measurement distinguishing these two structure type is the C1-C3 distance. Table 1 updates Table 5.11 from my book with the computed value of this distance using some new methods. In particular, the state-specific multireference coupled cluster Mk-MRCCSD method1 with the cc-pCVTZ basis set indicates a distance of 2.014 Å.2 The density cumulant functional theory3 ODC-124 with the cc-pCVTZ basis set also predicts the single ring structure with a distance of 2.101 Å.5
Table 1. C1-C3 distance (Å) with different computational methods using the cc-pCVTZ basis set
|
method |
r(C1-C3) |
|
CCSD5 |
1.556 |
|
CCSD(T)5 |
2.043 |
|
OCD-125 |
2.101 |
|
Mk-MRCCSD2 |
2.014 |
References
(1) Evangelista, F. A.; Allen, W. D.; Schaefer III, H. F. "Coupling term derivation and general implementation of state-specific multireference coupled cluster theories," J. Chem. Phys 2007, 127, 024102-024117, DOI: 10.1063/1.2743014.
(2) Jagau, T.-C.; Prochnow, E.; Evangelista, F. A.; Gauss, J. "Analytic gradients for Mukherjee’s multireference coupled-cluster method using two-configurational self-consistent-field orbitals," J. Chem. Phys. 2010, 132, 144110, DOI: 10.1063/1.3370847.
(3) Kutzelnigg, W. "Density-cumulant functional theory," J. Chem. Phys. 2006, 125, 171101, DOI: 10.1063/1.2387955.
(4) Sokolov, A. Y.; Schaefer, H. F. "Orbital-optimized density cumulant functional theory," J. Chem. Phys. 2013, 139, 204110, DOI: 10.1063/1.4833138.
(5) Mullinax, J. W.; Sokolov, A. Y.; Schaefer, H. F. "Can Density Cumulant Functional Theory Describe Static Correlation Effects?," J. Chem. Theor. Comput. 2015, 11, 2487-2495, DOI: 10.1021/acs.jctc.5b00346.
InChIs
1a: InChI=1S/C6H4/c1-2-4-6-5-3-1/h1-3,6H