A note here on a few recent Angew. Chem. articles of interest to readers of this blog. The first is a comment by Frenking1 concerning the “trilogue” by Shaik, Hoffmann and Rzepa2 which discusses the nature of C2, especially the notion that this molecules may possess a quadruple bond (see this post for a previous post on this article.) Frenking argues that the force constant associated with the C-C stretch in C2 is smaller than that in acetylene, so how can one argue that there is some quadruple bond character in C2? A reply from the original authors3 accompanies the comment by Frenking, and they respond by noting that the PES for bond stretching is unusually flat. I had the generally sense, though, that the authors of both articles were really talking past each other and that an opportunity for a more fruitful discussion has been missed.
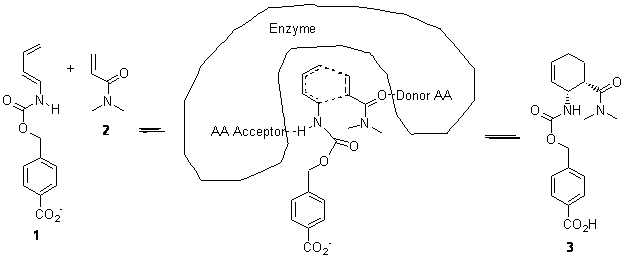
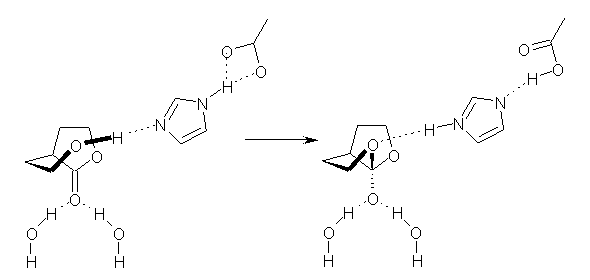
The other article of note is an excellent review of de novo enzyme design as performed by the Baker and Houk labs.4 This review, authored by leaders of this effort, highlights their approach to this “holy grail” problem. The general notion is to use standard tools of computational chemistry to design a theozyme. Next, this theozyme is placed into known protein motifs with the attempt to have it fit without too much steric clash. The protein is then mutated one residue at a time to optimize the fit and binding of the theozyme to substrate. Lastly, the best targets are synthesized and tested. (The reader can see my post one of their projects: synthetic Diels-Alderase.)
References
(1) Frenking, G.; Hermann, M. "Critical Comments on “One Molecule, Two Atoms, Three
Views, Four Bonds?”," Angew. Chem. Int. Ed. 2013, 52, 5922-5925. DOI: 10.1002/anie.201301485.
(2) Shaik, S.; Rzepa, H. S.; Hoffmann, R. "One Molecule, Two Atoms, Three Views, Four
Bonds?," Angew. Chem. Int. Ed. 2013, 52, 3020-3033, DOI: 10.1002/anie.201208206.
(3) Danovich, D.; Shaik, S.; Rzepa, H. S.; Hoffmann, R. "A Response to the Critical Comments on “One Molecule, Two Atoms, Three Views, Four Bonds?”," Angew. Chem. Int. Ed. 2013, 52, 5926-5928, DOI: 10.1002/anie.201302350.
(4) Kiss, G.; Çelebi-Ölçüm, N.; Moretti, R.; Baker, D.; Houk, K. N. "Computational Enzyme Design," Angew. Chem. Int. Ed. 2013, 52, 5700-5725, DOI: 10.1002/anie.201204077.