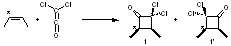Click chemistry has been used in a broad range of applications. The use of metal catalysts has limited its application to biological system, but the development of strain-promoted cycloaddition to cyclooctyne has opened up click chemistry to bioorthogonal labeling.
An interesting variation on this is the use of 1,2-benzoquinone 1 and substituted analogues as the Diels-Alder diene component. Escorihuela and co-workers have reported on the use of this diene with a number of cyclooctyne derivatives, measuring kinetics and also using computations to assess the mechanism.1
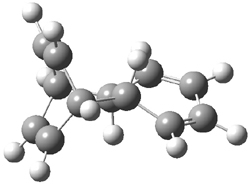
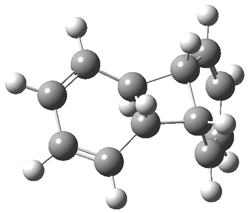
Their computations focused on two reactions using cyclooctyne 2 and the cyclopropane-fused analogue 3:
|
|
Reaction 1 |
|
|
Reaction 2 |
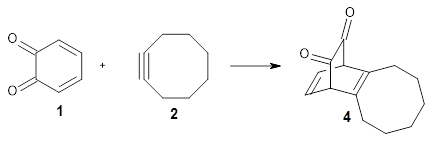
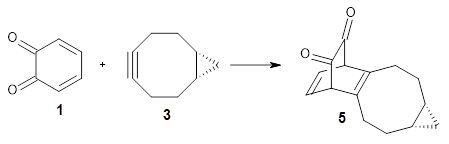
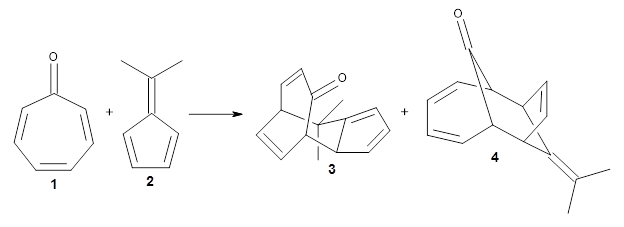
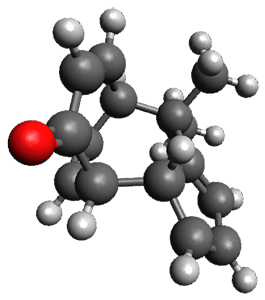
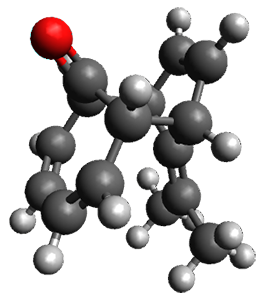
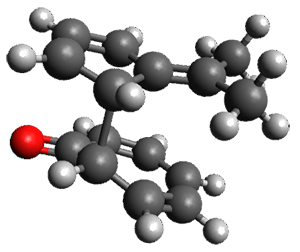
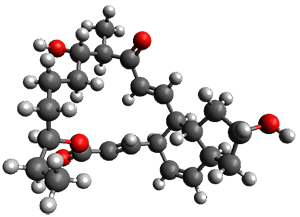
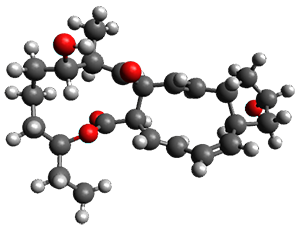
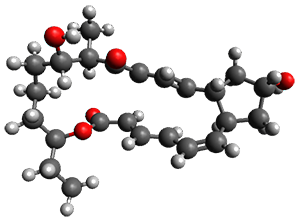
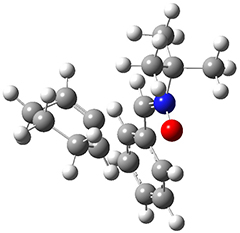
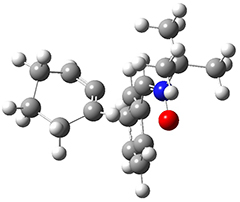
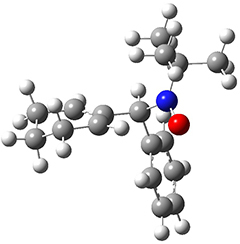
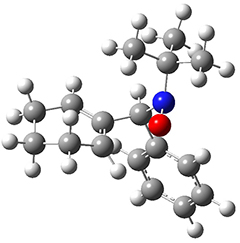
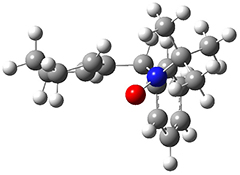
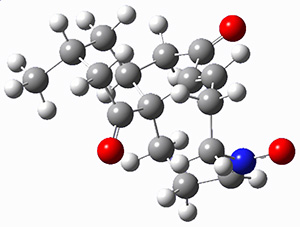
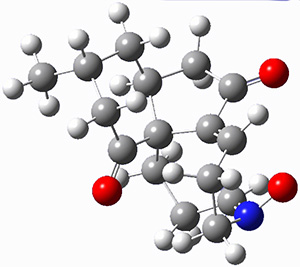
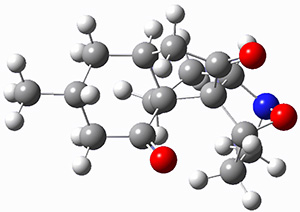
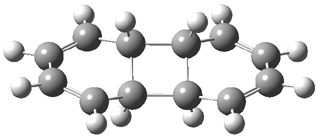
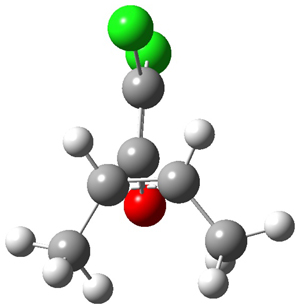
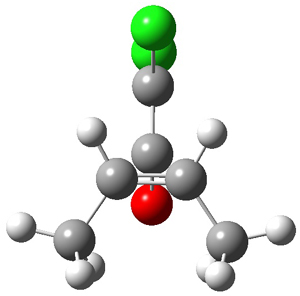
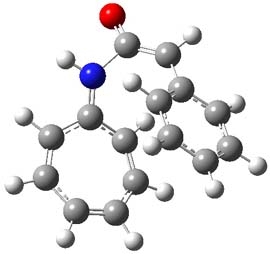
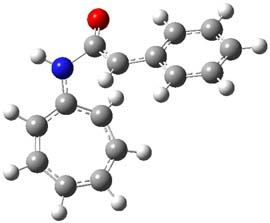
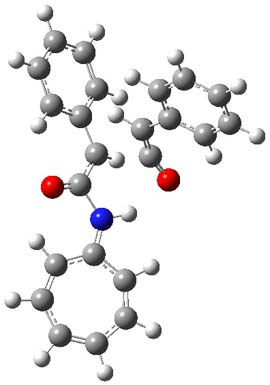
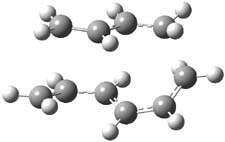
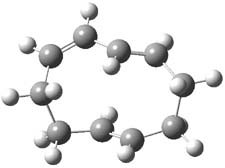
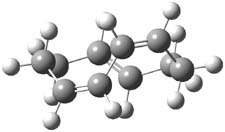
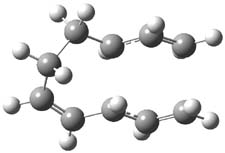
They examined these reactions with a variety of density functionals along with some post-HF methods. The transition states of the two reactions are shown in Figure 1. A variety of different density functionals and MP2 are consistent in finding synchronous or nearly synchronous transition states.
|
|
|
|
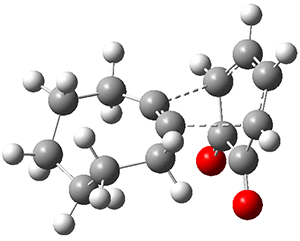
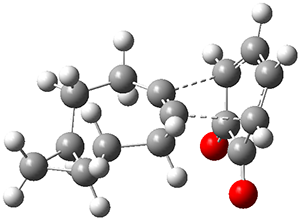
Figure 1. B97D/6-311+G(d,p) transition states for Reactions 1 and 2.
In terms of activation energies, all of the DFT methods consistently overestimate the barrier by about 5-10 kcal mol-1, with B97D-D3 doing the best. MP2 drastically underestimates the barriers, though the SOS-MP2 or SCS-MP2 improve the estimate. Both CCSD(T) and MR-AQCC provide estimates of about 8.5 kcal mol-1, still 3-4 kcal mol-1 too high. The agreement between CCSD(T), a single reference method, and MR-AQCC, a multireference method, indicate that the transition states have little multireference character. Given the reasonable estimate of the barrier afforded by B97D-D3, and its tremendous performance advantage over SCS-MP2, CCSD(T) and MR-AQCC, this is the preferred method (at least with current technology) for examining Diels-Alder reactions like these, especially with larger molecules.
References
1) Escorihuela, J.; Das, A.; Looijen, W. J. E.; van Delft, F. L.; Aquino, A. J. A.; Lischka, H.; Zuilhof, H., "Kinetics of the Strain-Promoted Oxidation-Controlled Cycloalkyne-1,2-quinone Cycloaddition: Experimental and Theoretical Studies." J. Org. Chem. 2018, 83, 244-252, DOI: 10.1021/acs.joc.7b02614.
InChIs
1: InChI=1S/C6H4O2/c7-5-3-1-2-4-6(5)8/h1-4H
InChIKey=WOAHJDHKFWSLKE-UHFFFAOYSA-N
2: InChI=1S/C8H12/c1-2-4-6-8-7-5-3-1/h1-6H2
InChIKey=ZPWOOKQUDFIEIX-UHFFFAOYSA-N
3: InChI=1S/C9H12/c1-2-4-6-9-7-8(9)5-3-1/h8-9H,3-7H2
InChIKey=rQDNSAFCVPAMWCJ-UHFFFAOYSA-N
4: InChI=1S/C14H16O2/c15-13-11-7-8-12(14(13)16)10-6-4-2-1-3-5-9(10)11/h7-8,11-12H,1-6H2
InChIKey=OQMYZEFKUMPECV-UHFFFAOYSA-N
5: InChI=1S/C15H16O2/c16-14-12-5-6-13(15(14)17)11-4-2-9-7-8(9)1-3-10(11)12/h5-6,8-9,12-13H,1-4,7H2/t8-,9+,12?,13?
InChIKey=NKDGTIVNLDJQKR-RFZWMSCOSA-N