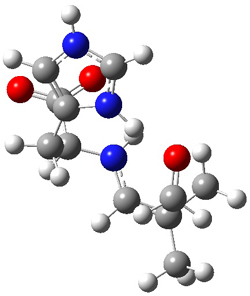
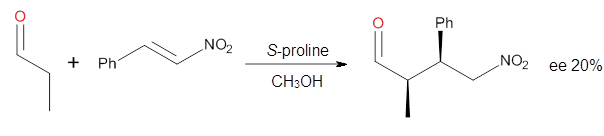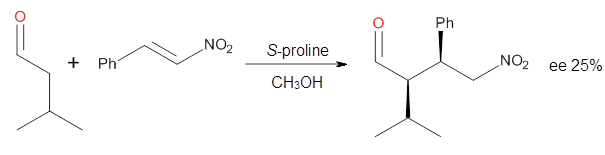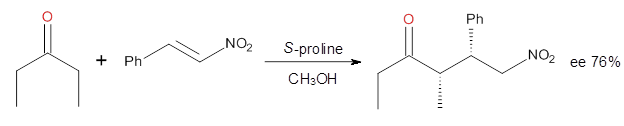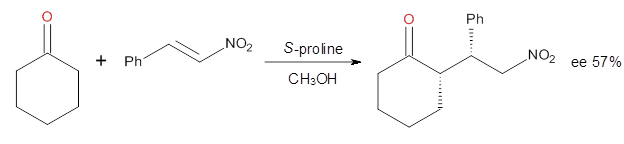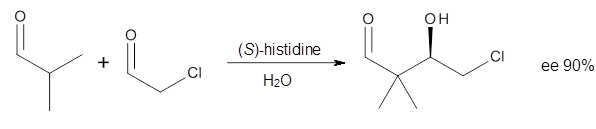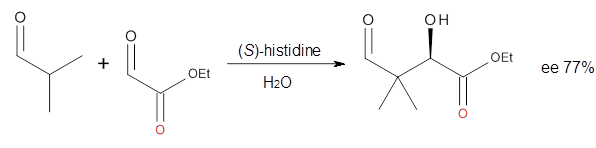How should one assess the aromatic stabilization energy of non-planar compounds like corannulene 1? The standard approach might be to employ the homodesmotic reaction, like Reaction 1. The energy of this reaction is however quite different whether one chooses s-cis or s-trans butadiene: 67.5 kcal mol-1 with the former and 14.8 kcal mol-1 with the latter. Exactly how one balances the total number of cis/trans relationships is problematic, but worse still is that Reaction 1 does not remove the effect of strain and non-planarity of 1.
|
|
|
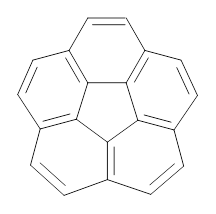
Reaction 1
15 H2C=CH-CH=CH2 + 20 H2C=C(CH3)2 → 1 + 10 H2C=CH2 + 20 H2C=CH-CH3
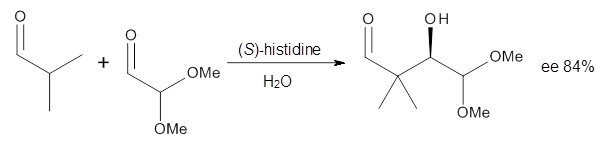
Dobrowolski, Ciesielski and Cyranski1 propose a series of reactions that extend the isomerization stabilization energy concept of Schleyer. References are chosen that involve fixed alternate polyenes by appending methylene groups, creating radialene-like compounds. Reaction 2 and 3 are two such reactions that attempt to remove strain and non-planarity effects along with balancing the cis/trans relationships and potential H-H clashes between the pendant methylene groups. They report an additional 18 variations, because there is no unique method for portioning these effects.
Reaction 2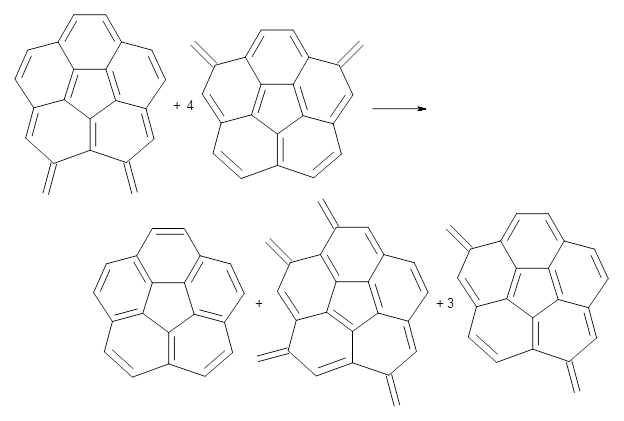
Reaction 3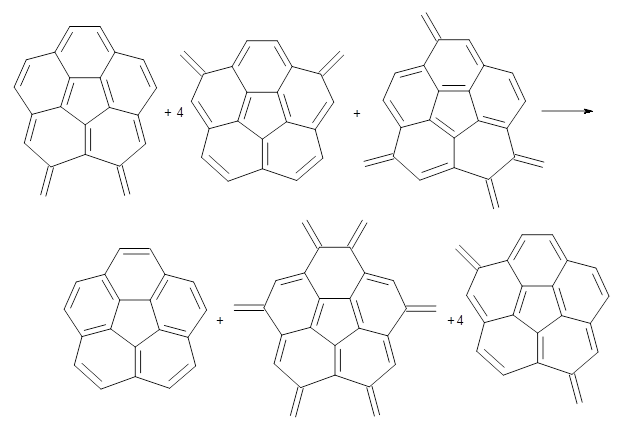
Using B3LYP/6-311G** energies with zero-point vibrational energy, the reaction energies are 46.7 and 46.3 kcal mol-1 for Reactions 2 and 3, respectively. Using all of the variations, the mean value is 44.7 kcal mol-1 with a standard deviation of only 1.2 kcal mol-1. It is clear that corranulene has a rather substantial artomatic stabilization energy, reflecting its decided aromatic character.
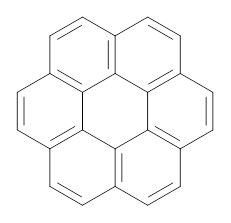
In a similar vein, they have also estimated the aromatic stabilization energy of coronene 2 as 58.4 kcal mol-1, which, while clearly demonstrating the 2 is aromatic, it does not express any “superaromaticity”.
References
(1) Dobrowolski, M. A.; Ciesielski, A.; Cyranski, M. K. "On the aromatic stabilization of corannulene and coronene," Phys. Chem. Chem. Phys., 2011, 13, 20557-20563, DOI: 10.1039/C1CP21994D
InChIs
1: InChI=1S/C21H14/c1-3-13-6-7-15-10-11-16-9-8-14-5-4-12(2)17-18(13)20(15)21(16)19(14)17/h3-11H,1H2,2H3
InChIKey=ZJQHTVPYWDRMLD-UHFFFAOYSA-N
2: InChI=1S/C24H12/c1-2-14-5-6-16-9-11-18-12-10-17-8-7-15-4-3-13(1)19-20(14)22(16)24(18)23(17)21(15)19/h1-12H
InChIKey=VPUGDVKSAQVFFS-UHFFFAOYSA-N