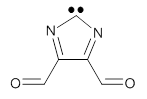
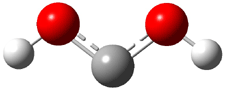
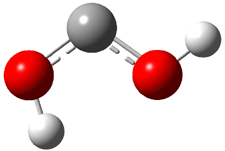
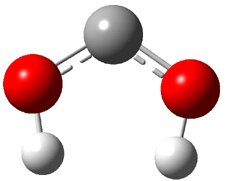
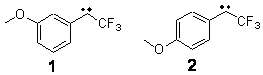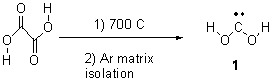The Schreiner group has again reported an amazing experimental and computational study demonstrating a fascinating quantum mechanical tunneling effect, this time for the trifluoromethylhydroxycarbene (CF3COH) 2.1 (I have made on a number of posts discussing a series of important studies in this field by Schreiner.) Carbene 2 is formed, in analogy to many other hydroxycarbenes, by flash vapor pyrolysis of the appropriate oxoacid 1 and capturing the products on a noble gas matrix.
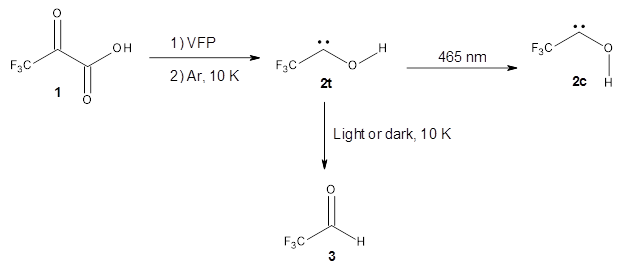
Carbene 2t is observed by IR spectroscopy, and its structure is identified by comparison with the computed CCSD(T)/cc-pVTZ frequencies. When 2t is subjected to 465 nm light, the signals for 2t disappear within 30s, and two new species are observed. The first species is the cis conformer 2c, confirmed by comparison with its computed CCSD(T)/cc-pVTZ frequencies. This cis conformer remains even with continued photolysis. The other product is determined to be trifluoroacetaldehyde 3. Perhaps most interesting is that 2t will convert to 3 in the absence of light at temperatures between 3 and 30 K, with a half-life of about 144 h. There is little rate difference at these temperatures. These results are quite indicative of quantum mechanical tunneling.
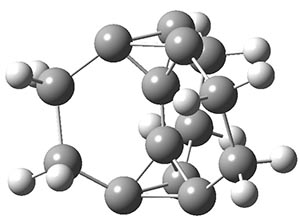
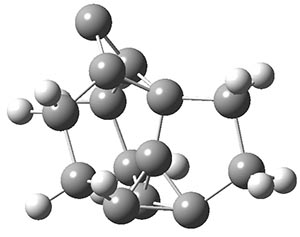
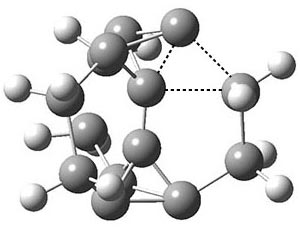
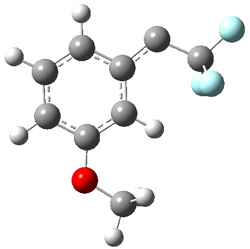
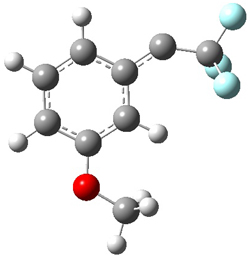
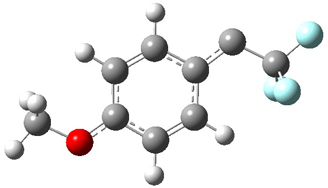
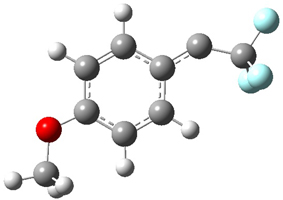
To aid in confirming tunneling, they computed the potential energy surface at CCSD(T)/cc-pVTZ. The trans isomer is 0.8 kcal mol-1 lower in energy that the cis isomer, and this is much smaller than for other hydroxycarbenes they have examined. The rotational barrier TS1 between the two isomer is quite large, 26.4 kcal mol-1, precluding their interchange by classical means at matrix temperatures. The barrier for conversion of 2t to 3 (TS2) is also quite large, 30.7 kcal mol-1, and insurmountable at 10K by classical means. No transition state connecting 2c to 3 could be located. These geometries and energies are shown in Figure 1.
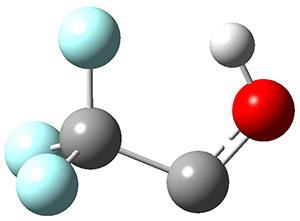
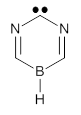
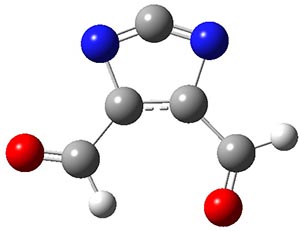
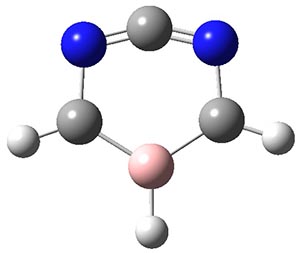
2c |
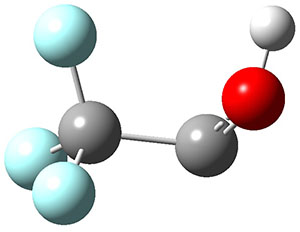
TS1 |
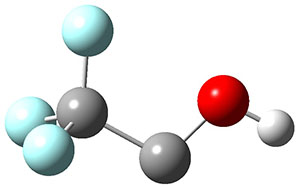
2t |
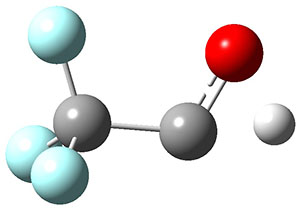
TS2 |
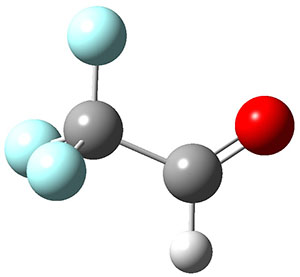
3 |
Figure 1. Optimized geometries at CCSD(T)/cc-pVTZ. Relative energies (kcal mol-1) of each species are listed as well.
WKB computations at M06-2X/6-311++G(d,p) predict a half-life of 172 h, in nice agreement with experiment. The computed half-life for deuterated 2t is 106 years, and the experiment on the deuterated analogue revealed no formation of deuterated 3.
The novel component of this study is that tunneling is conformationally selective. The CF3 group stabilizes the cis form probably through some weak H…F interaction, so that the cis isomer can be observed, but no tunneling is observed from this isomer. Only the trans isomer has the migrating hydrogen atom properly arranged for a short hop over to the carbon, allowing the tunneling process to take place.
References
1) Mardyukov, A.; Quanz, H.; Schreiner, P. R., "Conformer-specific hydrogen atom tunnelling in trifluoromethylhydroxycarbene." Nat. Chem. 2017, 9, 71–76, DOI: 10.1038/nchem.2609.
InChIs
1: =1S/C3HF3O3/c4-3(5,6)1(7)2(8)9/h(H,8,9)
InChIKey=GVDJEHMDNREMFA-UHFFFAOYSA-N
2: InChI=1S/C2HF3O/c3-2(4,5)1-6/h6H
InChIKey=FVJVNIREIXAWKU-UHFFFAOYSA-N
3: InChI=1S/C2HF3O/c3-2(4,5)1-6/h1H
InChIKey=JVTSHOJDBRTPHD-UHFFFAOYSA-N