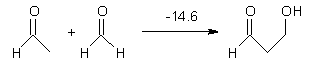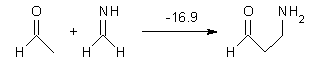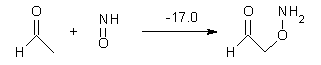The nature of reactions of indolynes is the subject of two recent computational/experimental studies. There are three isomeric indolynes 1a-c which are analogues of the more famous benzyne (which I discuss in significant detail in Chapter 4.4 of my book).

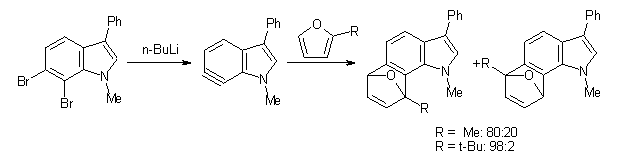
One might anticipate that the indolynes undergo comparable reactions as benzyne, like Diels-Alder reactions and nucleophilic attack. In fact the indolynes do undergo these reactions, with unusual regiospecificity. For example, the reaction of the substituted 6,7-indolyne undergoes regioselective Diels-Alder cycloaddition with substituted furans (Scheme 1), but the reaction with the other indolynes gives no regioselection. 1 Note that the preferred product is the more sterically congested adduct.
Scheme 1
In the case of nucleophilic addition, the nucleophiles add specifically to C6 with substituted 6,7-indolynes (Scheme 2), while addition to 4,5-indolynes preferentially gives the C5-adduct (greater than 3:1) while addition to the 5,6-indolynes preferentially gives the C5-adduct), but with small selectivity (less than 3:1).2
Scheme 2
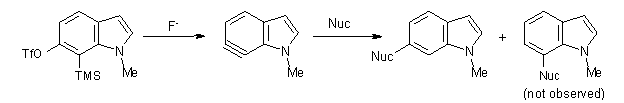
The authors of both papers – Chris Cramer studied the Diels-Alder chemistry and Ken Houk studied the nucleophilic reactions – employed DFT computations to examine the activation barriers leading to the two regioisomeric products. So for example, Figure 1 shows the two transition states for the reaction of 2c with 2-iso-propyl furan computed at MO6-2X/6-311+G(2df,p).
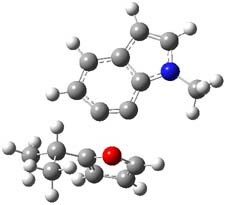 ΔG‡ = 9.7 |
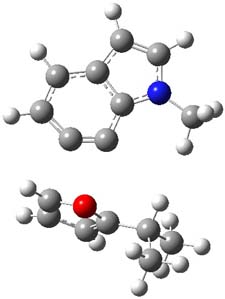 ΔG‡ = 7.6 |
Figure 1. MO6-21/6-311+G(2df,p) optimized TSs for the reaction of 2-iso-propylfuran with 2c. Activation energy (kcal mol-1) listed below each structure.1
The computational results are completely consistent with the experiments. For the Diels-Alder reaction of 2-t-butylfuran with the three indolynes 2a-c, the lower computed TS always corresponds with the experimentally observed major product. The difference in the energy of the TSs leading to the two regioisomers for reaction with 2a and 2b is small (less than 1 kcal mol-1), consistent with the small selectivity. On the other hand, no barrier could be found for the reaction of 2-t-butylfuran with 2c that leads to the major product. Similar results are also obtained for the nucleophilic addition – in all cases, the experimentally observed major product corresponds with the lower computed activation barrier.
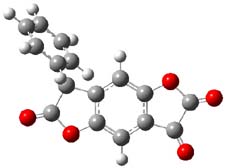
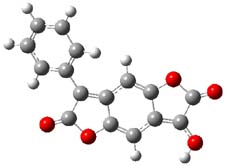
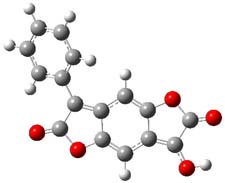
So what accounts for the regioselectivity? Both papers make the same argument, though couched in slightly different terms. Houk argues in terms of distortion energy – the energy needed to distort reactants to their geometries in the TS. As seen in Figure 2, the benzyne fragment of 2a is distorted, with the C-C-C angle at C4 of 125° and at C5 of 129°. In the transition states, the angle at the point of nucleophilic attack widens. Since the angle starts out wider at C5, attack there is preferred, since less distortion is needed to achieve the geometry of the TS.
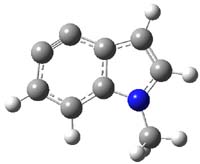 2a |
|
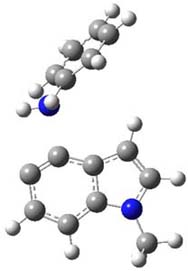 TS at C4 |
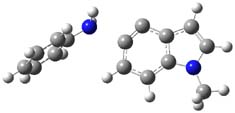 TS |
Figure 2. B3LYP/6-31G(d) optimized structures of 2a and the TSs for the reaction of aniline with 2a. Activation energy in kcal mol-1.2
Cramer argues in terms of the indolyne acting as an electrophile. Increasing substitution at the furan 2-position makes is better at stabilizing incipient positive charge that will build up there during a (very) asymmetric Diels-Alder transition state. This explains the increasing selectivity of the furan with increasing substitution. The indolyne acting as an electrophile means that the attack will lead from the center will lesser charge. In 2c, the C-C-C angle at C6 is 135.3°, while that at C7 is 117.2°. This makes C7 more carbanionic and C6 more carbocationic; therefore, the first bond made is to C6, leading to the more sterically congested product. Note that Houk’s argument applies equally well, as C6 is predistorted to the TS geometry.
References
(1) Garr, A. N.; Luo, D.; Brown, N.; Cramer, C. J.; Buszek, K. R.; VanderVelde, D., "Experimental and Theoretical Investigations into the Unusual Regioselectivity of 4,5-, 5,6-, and 6,7-Indole Aryne Cycloadditions," Org. Lett., 2010, 12, 96-99, DOI: 10.1021/ol902415s
(2) Cheong, P. H. Y.; Paton, R. S.; Bronner, S. M.; Im, G. Y. J.; Garg, N. K.; Houk, K. N., "Indolyne and Aryne Distortions and Nucleophilic Regioselectivites," J. Am. Chem. Soc., 2010, 132, 1267-1269, DOI: 10.1021/ja9098643
InChIs
1a: InChI=1/C8H5N/c1-2-4-8-7(3-1)5-6-9-8/h2,4-6,9H
InChIKey=RNDHGGYOIRREHC-UHFFFAOYAU
1b: InChI=1/C8H5N/c1-2-4-8-7(3-1)5-6-9-8/h3-6,9H
InChIKey=WWZQFJXNXMIWCD-UHFFFAOYAO
1c: InChI=1/C8H5N/c1-2-4-8-7(3-1)5-6-9-8/h1,3,5-6,9H
InChIKey=UHIRLIIPIXHWLT-UHFFFAOYAH
2a: InChI=1/C9H7N/c1-10-7-6-8-4-2-3-5-9(8)10/h3,5-7H,1H3
InChIKey=VTVUPAJGRVFCKI-UHFFFAOYAJ
2b: InChI=1/C9H7N/c1-10-7-6-8-4-2-3-5-9(8)10/h4-7H,1H3
InChIKey=KKPOWDDYMOXTFW-UHFFFAOYAN
2c: InChI=1/C9H7N/c1-10-7-6-8-4-2-3-5-9(8)10/h2,4,6-7H,1H3
InChIKey=MDAHOGWZOBLIEX-UHFFFAOYAZ