If you hadn’t noticed, I am a big fan of the work that Dan Singleton is doing concerning the role of dynamics in discerning reaction mechanisms. Dan’s group has reported another outstanding study combining experiments, traditional QM computations, and molecular dynamics – this time on the Wittig reaction.1
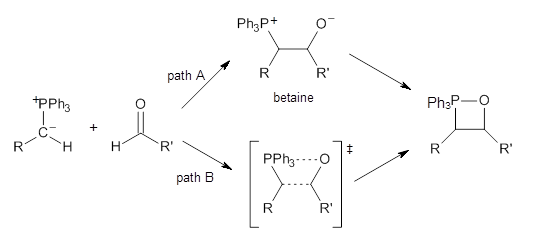
The key question concerning the mechanism is whether a betaine intermediate is accessed along the reaction (path A) or whether the reaction proceeds in a concerted manner (path B). Earlier computations had supported the concerted pathway (B).
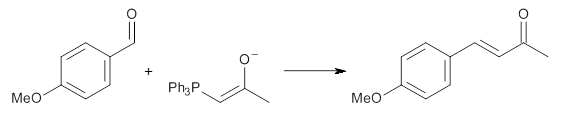
Experimental determination of the heavy atom kinetic isotope effect was made for Reaction 1.
Reaction 1
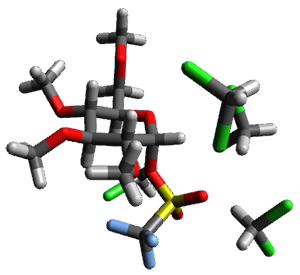
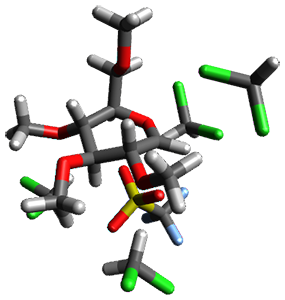
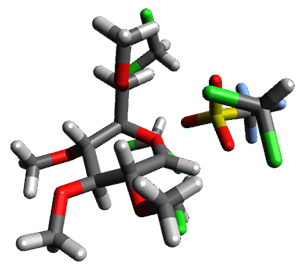
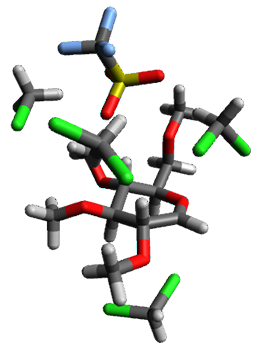
Using the 6-31+G(2df,p) basis set, three different density functionals predict three different potential energy surfaces. With M06-2x, the surface indicates path A (stepwise), with the first step rate-limiting. B3P86 also predicts the stepwise reaction, but the second step is rate-limiting. The Lc-wPBE functional predicts a concerted reaction. Using these surfaces, they predicted the carbon isotope effect and compared it to the experimental values. The best agreement is with the M06-2x surface with a weighting of the vibrational energies of the two different TSs. The optimized structures of the two transition states, the betaine intermediate, and the product are shown in Figure 1.
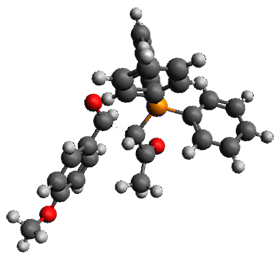
TS1 |
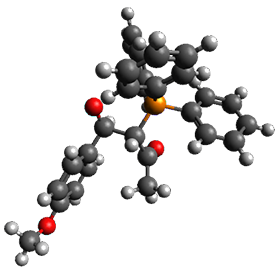
Betaine |
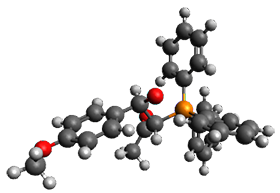
TS2 |
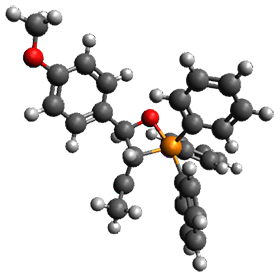
Product |
Figure 1. M06-2x/6-31+G(2df,p) optimized geometry of the critical points of Reaction 1.
The agreement of the predicted and experimental KIE is not ideal. So, they performed molecular dynamics computations with the ONIOM approach using M06-2x/6-31G* for Reaction 1 and 53 THF molecules treated at PM3. 360 trajectories were begun in the region of the first transition state (TS1), and they can be organized into 4 groups. The first group (128 trajectories) are reactions that produce product. The second group (76 cases) form the C-C bond but then it ruptures and returns to reactant. The third group (82 cases) have an immediate recrossing back to reactant, and the last group (16 cases) takes product back to the first TS and then returns to product. The predicted KIE using this weighted MD results gives values in outstanding agreement with the experiments.
Of the first group, about 50% pass from TS1 to TS2 in less than 150 fs, or in other words look like a concerted path. But a good number of trajectories reside in the betaine region for 1-2 ps.
In contrast, trajectories initiated from the betaine with equilibrated THF molecules indicate a median of 600 ps to travel from TS1 to TS2 and do not resemble a concerted path.
They argue that this bimodal distribution is in part associated with a solvent effect. When the first TS is crossed the solvent molecules are not equilibrated about the solute, and 10-20% of the trajectories immediately pass through the betaine region due to “dynamic matching” where the entering motion matches with exiting over the second transition state. The longer trajectories result from improper dynamic matching, but faster motion in the solute than motion amongst the solvent needed to stabilize the betaine. So, not only do we need to be concerned about dynamic effects involving the reactants, we need to be concerned about dynamics associated with the solvent too!
References
(1) Chen, Z.; Nieves-Quinones, Y.; Waas, J. R.; Singleton, D. A. "Isotope Effects, Dynamic Matching, and Solvent Dynamics in a Wittig Reaction. Betaines as Bypassed Intermediates," J. Am. Chem. Soc. 2014, 136, 13122-13125, DOI: 10.1021/ja506497b.