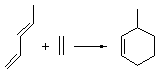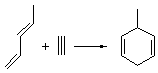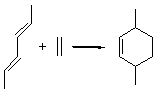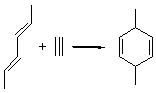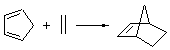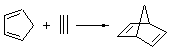Singleton has taken another foray into the murky arena of “dynamic effects”, this time with the aim of trying to provide some guidance towards making qualitative product predictions.1 He has examined four different Diels-Alder reaction involving two diene species, each of which can act as either the diene or dienophile. I will discuss the results of two of these reactions, namely the reactions of 1 with 2 (Reaction 1) and 1 with 3 (Reaction 2).
|
Reaction 1

|
|
Reaction 2

|
In the experimental studies, Reaction 1 yields only 4, while reaction 2 yields both products in the ratio 6:7 = 1.6:1. Standard transition state theory would suggest that there are two different transition states for each reaction, one corresponding to the 4+2 reaction where 1 is the dienophile and the other TS has 1 as the dienophile. Then one would argue that in Reaction 1, the TS leading to 4 is much lower in energy than that leading to 5, and for Reaction 2, the TS state leading to 6 lies somewhat lower in energy than that leading to 7.
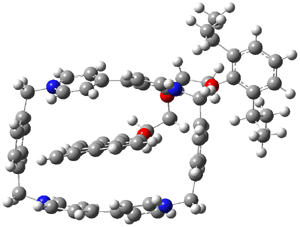
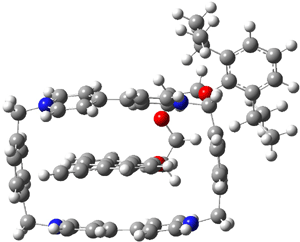
Now the interesting aspect of the potential energy surfaces for these two reactions is that there are only two transition states. The first corresponds to the Cope rearrangement between the two products (connecting 4 to 5 on the PES of Reaction 1 and 6 to 7 on the PES of Reaction 2). That leaves only one TS connecting reactants to products! These four TSs are displayed in Figure 1.
|
Reaction 1
|
Reaction 2
|
|
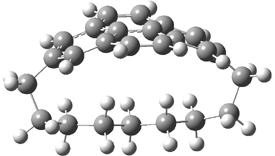
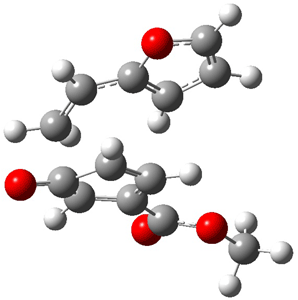
TS 12→45
|
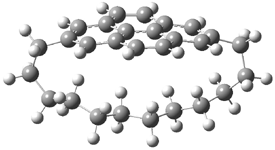
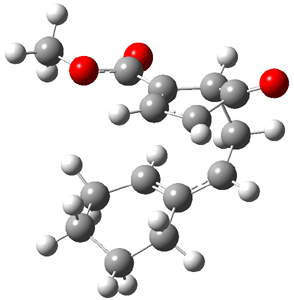
TS 13→67
|
|
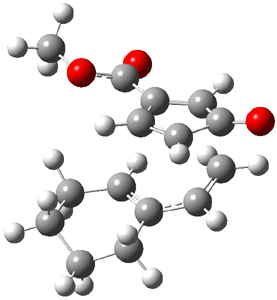
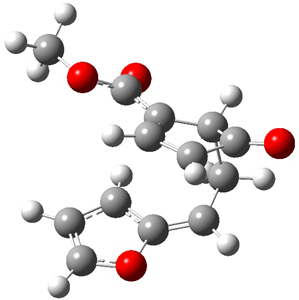
Cope TS 4→5
|
Cope TS 6→7
|
Figure 1. MPW1K/6-31+G** TSs on the PES of Reactions 1 and 2.1
These transition states are “bispericyclic” (first recognized by Caramella2), having the characteristics of both possible Diels-Alder reactions, i.e. for Reaction 1 these are the [4π1+2π2] and [4π2+2π1]. What this implies is that the reactants come together, cross over a single transition states and then pass over a bifurcating surface where the lowest energy path (the IRC or reaction path) continues on to one product only. The second product, however, can be reached by passing over this same transition state and then following some other non-reaction path. This sort of surface is ripe for experiencing non-statistical behavior, or “dynamic effects”.
Trajectory studies were then performed to explore the product distributions. Starting from TS 12→45, 39 trajectories were followed: 28 ended with 4 and 10 ended with 5 while one trajectory recrossed the transition state. Isomerization of 5 into 4 is possible, and the predicted low barrier for this explains the sole observation of 4. For Reaction 2, of the 33 trajectories that originated at TS 13→67, 12 led to 6 and 19 led to 7. This distribution is consistent with the experimental product distribution of a slight excess of 7 over 6.
Once again we see here a relatively simple reaction whose product distribution is only interpretable using expensive trajectory computations, and the result leave little simplifying concepts to guide us in generalizing to other (related) systems. Singleton does provide two rules-of-thumb that may help prod us towards creating some sort of dynamic model. First, he notes that the geometry of the single transition state that “leads” to the two products can suggest the major product. The TS geometry can be “closer” to one product over the other. For example, in TS 12→45 the two forming C-C bonds that differentiate the two products are 2.95 and 2.99 Å, and the shorter distance corresponds to forming 4. In TS 13→67, the two C-C distances are 2.83 and 3.13 Å, with the shorter distance corresponding to forming 6. The second point has to do with the position of the second TS, the one separating the two products. This TS acts to separate the PES into two basins, one for each product. The farther this TS is from the first TS, the greater the selectivity.
As Singleton notes, neither of these points is particularly surprising in hindsight. Nonetheless, since we have so little guidance in understanding reactions that are under dynamic control, any progress here is important.
References
(1) Thomas, J. B.; Waas, J. R.; Harmata, M.; Singleton, D. A., "Control Elements in Dynamically Determined Selectivity on a Bifurcating Surface," J. Am. Chem. Soc. 2008, 130, 14544-14555, DOI: 10.1021/ja802577v.
(2) Caramella, P.; Quadrelli, P.; Toma, L., "An Unexpected Bispericyclic Transition Structure Leading to 4+2 and 2+4 Cycloadducts in the Endo Dimerization of Cyclopentadiene," J. Am. Chem. Soc. 2002, 124, 1130-1131, DOI: 10.1021/ja016622h
InChIs
1: InChI=1/C7H6O3/c1-10-7(9)5-2-3-6(8)4-5/h2-4H,1H3
InChIKey=XDEAUYSKQHEYSC-UHFFFAOYAM
2: InChI=1/C8H12/c1-2-8-6-4-3-5-7-8/h2,6H,1,3-5,7H2
InChIKey=SDRZFSPCVYEJTP-UHFFFAOYAI
3: InChI=1/C6H6O/c1-2-6-4-3-5-7-6/h2-5H,1H2
InChIKey=QQBUHYQVKJQAOB-UHFFFAOYAO
4: InChI=1/C15H18O3/c1-18-14(17)15-9-8-13(16)12(15)7-6-10-4-2-3-5-11(10)15/h6,8-9,11-12H,2-5,7H2,1H3/t1,12-,15+/m1/s1
InChIKey=IASNDVSMFFVIFJ-GDHFLIHABF
5: InChI=1/C15H18O3/c1-18-15(17)13-8-11-10(7-12(13)14(11)16)9-5-3-2-4-6-9/h5,8,10-12H,2-4,6-7H2,1H3
InChIKey=XOFSMKQRRVWZHS-UHFFFAOYAW
6: InChI=1/C13H12O4/c1-16-13(15)10-6-8-7(5-9(10)12(8)14)11-3-2-4-17-11/h2-4,6-9H,5H2,1H3
InChIKey=HTSLDILNKGZMHE-UHFFFAOYAH
7: InChI=1/C13H12O4/c1-16-12(15)13-6-4-10(14)8(13)2-3-11-9(13)5-7-17-11/h3-9H,2H2,1H3/t8-,9?,13-/m1/s1
InChIKey=URYPWPBQFGUBGW-KEJGKJRFBM