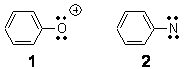Single-walled carbon nanotubes (SWNT) can be thought of as built from component macrocycles, often called nanohoops. So, for example, cycloparaphenylenes like 1 can be the thought of as the precursor (at least in principle) of armchair SWNTs. To create chiral SWNTs, Itami1 has suggested that cycloparaphenylene-naphthalene (2) and other acene substituted macrocycles would serve as appropriate precursors.
|
|
|
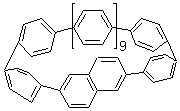
Itami has synthesized 2 (having 13 phenyl groups and one naphthyl group) and also examined the ring strain energy and racemization energy of a series of these types of compounds at B3LYP/6-31G(d). As might be expected, based on studies of the cycloparaphenylenes themselves,2,3 ring strain energy decreases with increasing size of the macrocycle. So, for example, the macrocycle with one naphthyl group and 5 phenyl rings has a strain energy of 90 kcal mol-1, but the strain is reduced to 40 kcal mol-1 with 13 phenyl rings.
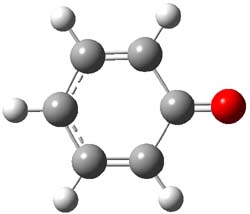
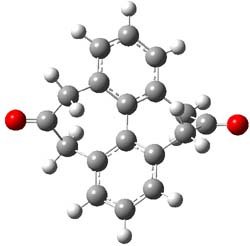
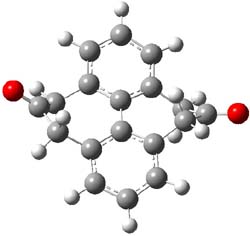
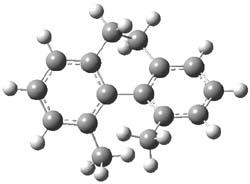
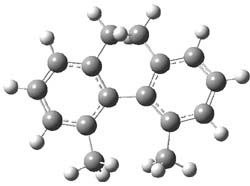
The macrocycle 2 and related structures are chiral, existing in P and M forms. The racemization involves first rotation of the naphthyl group, as shown in Figure 1, with a barrier of about 8 kcal mol-1. The direct product has the opposite stereochemistry but is not in the lowest energy conformation. Rotations of some phenyl groups remains to occur, but these rotations are expected to have a barrier less than that for the rotation of the naphthyl group, based on the previous study of cycloparaphenylenes. Again, the racemization barrier decreases with the size of the macrocycle.
 (P)-2 |
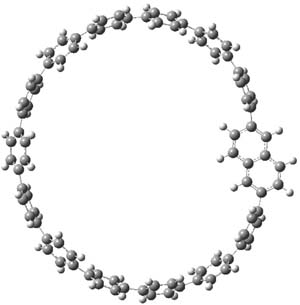 2-TS |
 (M)-2’ |
|
Figure 1. B3LYP/6-31G(d) optimized structures along the racemization pathway of 2.
References
(1) Omachi, H.; Segawa, Y.; Itami, K., "Synthesis and Racemization Process of Chiral Carbon Nanorings: A Step toward the Chemical Synthesis of Chiral Carbon Nanotubes," Org. Lett., 2011, 13, 2480-2483, DOI: 10.1021/ol200730m
(2) Segawa, Y.; Omachi, H.; Itami, K., "Theoretical Studies on the Structures and Strain Energies of Cycloparaphenylenes," Org. Lett., 2010, 12, 2262-2265, DOI: 10.1021/ol1006168
(3) Bachrach, S. M.; Stuck, D., "DFT Study of Cycloparaphenylenes and Heteroatom-Substituted Nanohoops," J. Org. Chem., 2010, 75, 6595-6604, DOI: 10.1021/jo101371m
InChIs
2: InChI=1/C88H58/c1-2-60-4-3-59(1)61-5-9-63(10-6-61)65-13-17-67(18-14-65)69-21-25-71(26-22-69)73-29-33-75(34-30-73)77-37-41-79(42-38-77)81-45-49-83(50-46-81)85-53-55-88-58-86(54-56-87(88)57-85)84-51-47-82(48-52-84)80-43-39-78(40-44-80)76-35-31-74(32-36-76)72-27-23-70(24-28-72)68-19-15-66(16-20-68)64-11-7-62(60)8-12-64/h1-58H/b61-59-,62-60-,65-63-,66-64-,69-67-,70-68-,73-71-,74-72-,77-75-,78-76-,81-79-,82-80-,85-83-,86-84+
InChIKey=VXOGKWSXPGSUSO-ZMOMEJFTBU