Peter Vollhardt and Ken Houk have teamed up on an interesting account of pericyclic reactions of molecules related to starphenylene.1 This touches on the nature of aromatic compounds and pericyclic reaction mechanisms, topics I take up in a few places in the book.
Compound 1 rearranges at 120 °C to 3, and the presumed pathway is
through 2 – the simultaneous [2+2+2] ring opening through the all-disrotatory path.
However, the computed (B3LYP/6-31G(d) activation energy is 34.6 kcal mol-1 for this path, much higher than the experimental activation enthalpy, which is 28.9 kcal mol-1.

The alternative path is to sequential break the cyclobutene rings with the standard conrotatory stereochemistry. This would give 4 and the barrier is 32.5 kcal mol-1, in better agreement with experiment. From here, there is a bond shift, which traverses a Möbius geometry – as proposed by Karney and Castro (see the book and also this previous post). An electrocylization, followed by a Diels-Alder cycloaddition completes the path to 3. The rate determining step is the first: 1 ↔ 4.
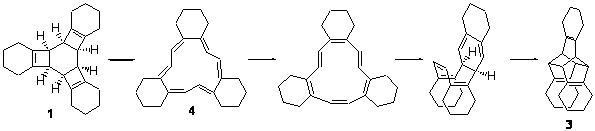
On the other hand, upon heating 5 produces 6. Here the computed barrier for the [2+2+2] reaction (32.6 kcal mol-1) is in nice agreement with the experimental value (34.1 kcal mol-1), while the stepwise pathway has a much higher barrier (39.9 kcal mol-1). They did not locate the polycyclic analogue of 3 (namely, 7) in the reaction of 5. This may be due in part to the fact that the bond shift is accompanied by a loss of aromaticity.

References
(1) Eichberg, M. J. H., K. N.; Lehmann, J.; Leonard, P. W.; Märker, A.; Norton, J. E.; Sawicka, D.; Vollhardt, K. P. C. W., G. D.; Wolff, S., "The Thermal Retro[2+2+2] cycloaddition of Cyclohexane Activated by Triscyclobutenannelation: Concerted All-Disrotatory versus Stepwise Conrotatory Pathways to Fused [12]Annulenes," Angew. Chem. Int. Ed., 2007, 46, 6894-6898, DOI: 10.1002/anie.200702474
InChIs
1: InChI=1/C24H30/c1-2-8-14-13(7-1)19-20(14)22-17-11-5-6-12-18(17)24(22)23-16-10-4-3-9-15(16)21(19)23/h19-24H,1-12H2/t19-,20+,21-,22+,23-,24+
2: InChI=1/C24H30/c1-2-8-20-15-16-22-10-5-6-12-24(22)18-17-23-11-4-3-9-21(23)14-13-19(20)7-1/h13-18H,1-12H2/b14-13-,16-15-,18-17-,19-13-,20-15+,21-14+,22-16+,23-17+,24-18-
3: InChI=1/C24H30/c1-2-8-14-13(7-1)19-21-15-9-3-4-10-16(15)22-20(14)23(19)17-11-5-6-12-18(17)24(21)22/h19-24H,1-12H2
4: InChI=1/C24H30/c1-2-8-20-15-16-22-10-5-6-12-24(22)18-17-23-11-4-3-9-21(23)14-13-19(20)7-1/h13-18H,1-12H2/b14-13+,16-15+,18-17+,19-13-,20-15+,21-14+,22-16-,23-17-,24-18+
5: InChI=1/C24H18/c1-2-8-14-13(7-1)19-20(14)22-17-11-5-6-12-18(17)24(22)23-16-10-4-3-9-15(16)21(19)23/h1-12,19-24H/t19-,20+,21-,22+,23-,24+
6: InChI=1/C24H18/c1-2-8-20-15-16-22-10-5-6-12-24(22)18-17-23-11-4-3-9-21(23)14-13-19(20)7-1/h1-18H/b14-13-,16-15-,18-17-,19-13-,20-15+,21-14+,22-16+,23-17+,24-18-
7: InChI=1/C24H18/c1-2-8-14-13(7-1)19-21-15-9-3-4-10-16(15)22-20(14)23(19)17-11-5-6-12-18(17)24(21)22/h1-12,19-24H