I guess one can never know enough about the SN2 reaction! Wester and co-workers have performed careful crossed molecular beam imagining on the reaction Cl– + CH3I.1 In collaboration with Hase, they have employed MP2/ECP/aug-cc-pVDZ computations to get the potential energy surface for the reaction and direct molecular dynamics. The PES is exactly as one would expect for a gas phase ion-molecule reaction: the transition state has backside attack of the nucleophile and it connects to two ion-dipole complexes (see Chapter 5.1.1).
The experiments are interpreted with the help of the MD computations. At low energy one sees formation of the complex. At higher energies, the direct backside attack reaction occurs. And at higher energies a new reaction path emerges, as sketched out in Figure 1. As the nucleophile (chloride) approaches methyl iodide, the methyl group rotates towards the nucleophile. The methyl group then collides with the nucleophile, which sends the methyl group spinning about the iodine atom in the opposite direction. The methyl group rotates all the way around the iodine atom and when it approaches the chloride a second time, the displacement reaction occurs and product is formed. They term this process a “roundabout mechanism”, and they have some experimental evidence for the occurrence of the double roundabout (two rotations of the methyl group about the iodine)! I think we should anticipate seeing more and more interesting reaction pathways as experimental and theoretical techniques continue to allow us a more detailed and precise view of motion of individual molecules across barriers.
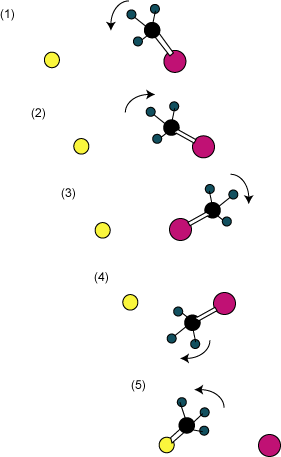
Figure 1. Schematic of the trajectory illustrating the roundabout mechanism.
Chlorine is yellow, iodine is pink and carbon is black.
References
(1) Mikosch, J.; Trippel, S.; Eichhorn, C.; Otto, R.; Lourderaj, U.; Zhang, J. X.; Hase, W. L.; Weidemüller, M.; Wester, R., "Imaging Nucleophilic Substitution Dynamics," Science 2008, 319, 183-186, DOI: 10.1126/science.1150238.