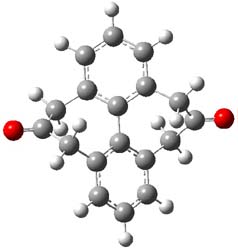
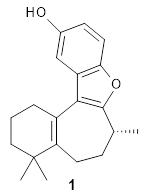
The structure of (+)-frondosin B 1 has been the subject of some concern. The compound has been synthesized by a number of research groups with the expected R isomer as the target. However, the Danishefsky1 and MacMillan2 synthesis led to a molecule with [α]D of about +16°, while Trauner3 reports a value of -16.8° and Ovaska4 prepared the S isomer with [α]D = -17.3°. Something is amiss here.
Joyce and coworkers have looked into this structure problem through a combination of advanced analytical techniques and computational chemistry.5 They utilize optical activity, electronic circular dichroism (ECD) and vibrational circular dichroism (VCD) and compare the experiments with computational results. IR and VCD were computed at B3LYP/6-31G** using a Boltzmann-weighted set of low-energy conformations. ECD computations were done at CAM-B3LYP/6-31++G**//B3LYP/6-31G**.
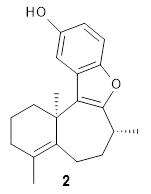
Basically, they found that (+)-frondosin B does have the R stereocenter. The different synthetic schemes did actually all lead to the same isomer, tested by looking at key intermediates along the way. The discrepancy in the optical activity is due to a small impurity, 2, that has the opposite rotation and a magnitude 10 times greater than that of authentic 1.
This paper is another nice example demonstrating the power of modern computational approaches to spectra that can be extremely valuable in structure determination. Organic chemists of all stripes should certainly be aware of how this tool can complement experiments.
My thanks to Derek Lowe who posted on this paper in his In The Pipeline blog.
References
1) Inoue, M.; Carson, M. W.; Frontier, A. J.; Danishefsky, S. J., "Total Synthesis and Determination of the Absolute Configuration of Frondosin B." J. Am. Chem. Soc.
2001, 123, 1878-1889, DOI: 10.1021/ja0021060.
2) Reiter, M.; Torssell, S.; Lee, S.; MacMillan, D. W. C., "The organocatalytic three-step total synthesis of (+)-frondosin B." Chem. Sci. 2010, 1, 37-42, DOI: 10.1039/C0SC00204F.
3) Hughes, C. C.; Trauner, D., "Palladium-catalyzed couplings to nucleophilic heteroarenes: the total synthesis of (−)-frondosin B." Tetrahedron 2004, 60, 9675-9686, DOI: 10.1016/j.tet.2004.07.041.
4) Ovaska, T. V.; Sullivan, J. A.; Ovaska, S. I.; Winegrad, J. B.; Fair, J. D., "Asymmetric Synthesis of Seven-Membered Carbocyclic Rings via a Sequential Oxyanionic 5-Exo-Dig Cyclization/Claisen Rearrangement Process. Total Synthesis of (−)-Frondosin B." Org. Letters 2009, 11, 2715-2718, DOI: 10.1021/ol900967j.
5) Joyce, L. A.; Nawrat, C. C.; Sherer, E. C.; Biba, M.; Brunskill, A.; Martin, G. E.; Cohen, R. D.; Davies, I. W., "Beyond optical rotation: what’s left is not always right in total synthesis." Chem. Sci. 2018, 9, 415-424, DOI: 10.1039/C7SC04249C.
InChIs
1: InChI=1S/C20H24O2/c1-12-6-8-16-14(5-4-10-20(16,2)3)18-15-11-13(21)7-9-17(15)22-19(12)18/h7,9,11-12,21H,4-6,8,10H2,1-3H3/t12-/m1/s1
InChIKey=LSPMJSWSYGOLFD-GFCCVEGCSA-N
2: InChI=1S/C20H24O2/c1-12-5-4-10-20(3)16(12)8-6-13(2)19-18(20)15-11-14(21)7-9-17(15)22-19/h7,9,11,13,21H,4-6,8,10H2,1-3H3/t13-,20-/m1/s1
InChIKey=ZBXZDKMLFIJFHG-ZUOKHONESA-N