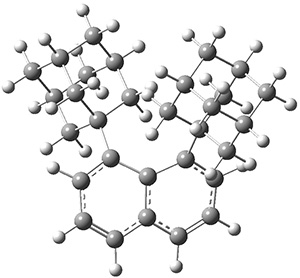
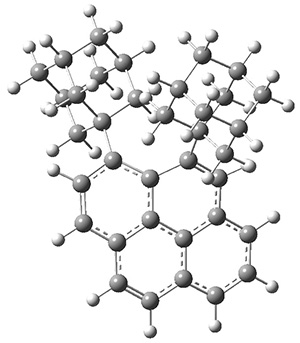
The notion of tunneling control has been a topic of interest within this blog a number of times. As developed by Schreiner and Allen,1,2 tunneling control is a third means for predicting (or directing) the outcome of a reaction, alongside the more traditionally recognized kinetic and thermodynamic control. Tunneling control occurs when tunneling through a higher barrier is preferred over tunneling through a lower barrier.
Kozuch and Borden propose another example of tunneling control, this time in the rearrangement of the noradamantyl carbene 1.3 This carbene can undergo a 1,2-carbon shift, driven by strain relief to form the alkene 2. The alternative as a 1,2-hydrogen shift that produces the alkene 3.
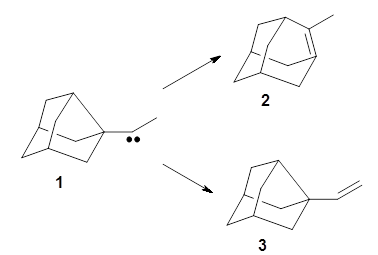
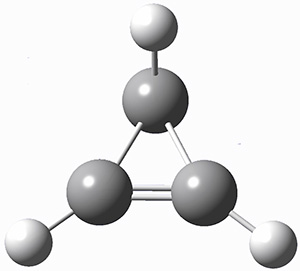
These two reaction pathways were explored using B3LYP/6-31G(d,p) computations coupled with canonical variational theory and small curvature tunneling corrections. Structures of the reactant 1 and the two transition states leading to the two products 2 and 3 are shown in Figure 1. The activation barrier at 300 K is 5.4 kcal mol-1 leading to 2 and 8.6 kcal mol-1 leading to 3. Tunneling is expected to be much more important for the hydrogen shift than for the carbon shift, but even including tunneling, the rate to form 2 is much faster than the rate to form 3 at 300 K.
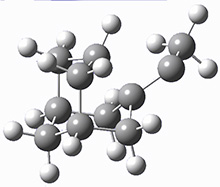 1 |
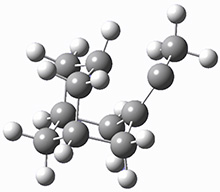 TS 1→2 |
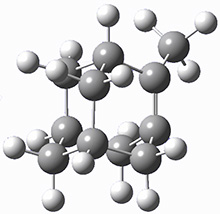 2 |
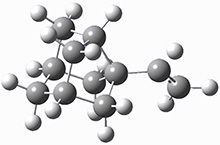 TS 1→3 |
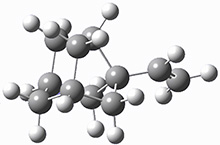 3 |
Figure 1. B3LYP/6 optimized structures of 1-3 and the transition states leading to 2 and 3.
The situation is reversed however at cryogenic temperatures (< 20 K). Tunneling is now the only route for the reactions to occur, and the rate for formation of 3 is dramatically greater than the rate of formation of 2, which is inhibited by the movement of the much heavier carbon atom. Perdeuteration of the methyl group of 1, which drastically slows the rate of tunneling in the path to 3, nonetheless still favors this pathway (forming d3–3) over formation of d3–2. Thus, at low temperatures the formation of 3 is the preferred product, a manifestation of tunneling control.
Kozuch and Borden end their paper with a hope that an experimentalist will examine this interesting case. I concur!
References
(1) Schreiner, P. R.; Reisenauer, H. P.; Ley, D.; Gerbig, D.; Wu, C.-H.; Allen, W. D. "Methylhydroxycarbene: Tunneling Control of a Chemical Reaction," Science 2011, 332, 1300-1303, DOI: 10.1126/science.1203761.
(2) Ley, D.; Gerbig, D.; Schreiner, P. R. "Tunnelling control of chemical reactions – the organic chemist’s perspective," Org. Biomol. Chem. 2012, 10, 3781-3790, DOI: 10.1039/C2OB07170C.
(3) Kozuch, S.; Zhang, X.; Hrovat, D. A.; Borden, W. T. "Calculations on Tunneling in the Reactions of Noradamantyl Carbenes," J. Am. Chem. Soc. 2013, 135, 17274-17277, DOI: 10.1021/ja409176u.
InChIs
1: InChI=1S/C11H16/c1-2-11-6-8-3-9(7-11)5-10(11)4-8/h8-10H,3-7H2,1H3
InChIKey=CXFJINASYYTBBV-UHFFFAOYSA-N
2: InChI=1S/C11H16/c1-7-10-3-8-2-9(5-10)6-11(7)4-8/h8-10H,2-6H2,1H3
InChIKey=XDANPUSLLJWVEK-UHFFFAOYSA-N
3: InChI=1S/C11H16/c1-2-11-6-8-3-9(7-11)5-10(11)4-8/h2,8-10H,1,3-7H2
InChIKey=JHEPVTWREMDEMG-UHFFFAOYSA-N