Here’s another nice example of computed NMR spectra being
used to identify complex organic structures.1
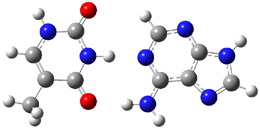
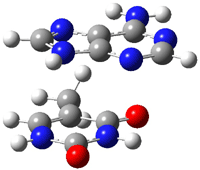
An alkaloid isolated from the custard apple tree was assigned the structure 1 and christened with the name samoquasine A.2 Two years later, the authors determined that samoquasine A was actually identical to perlolidine 2.3 Independent synthesis of the compound with structure 1 showed that its properties were not identical to that of samoquasine A.4,5 The properties of perlolidine were then found to differ from that of samoquasine A,4 leaving a void as to just what is the structure of samoquasine A.

Given that compounds 1 and the related compounds 3 and 4 had been prepared and their NMR spectra obtained, Timmons and Wipf1 decided to compute the 13C NMR spectra of 48 related compounds at B3LYP/6-311+G(2d,p)//B3LYP/6-31G(d). The mean absolute difference between the computed and experimental chemical shifts for 1, 3 and 4 are less than 2 ppm. Of the remaining 45 compounds, the one whose chemical shifts match best with that of samoquasine A is 2, with a mean absolute deviation of 1.8 ppm. This agreement supports the contention that samoquasine A and perlolidine are in fact identical. The authors contend that the experimental data used to conjecture that they were not identical is in fact faulty.
References
(1) Timmons, C.; Wipf, P., "Density Functional Theory Calculation of 13C NMR Shifts of Diazaphenanthrene Alkaloids: Reinvestigation of the Structure of Samoquasine A," J. Org. Chem., 2008, 73, 9168-9170, DOI: 10.1021/jo801735e.
(2) Morita, H.; Sato, Y.; Chan, K.-L.; Choo, C.-Y.; Itokawa, H.; Takeya, K.; Kobayashi, J. i., "Samoquasine A, a Benzoquinazoline Alkaloid from the Seeds of Annona squamosa," J. Nat. Prod., 2000, 63, 1707-1708, DOI: 10.1021/np000342i.
(3) Morita, H.; Sato, Y.; Chan, K.-L.; Choo, C.-Y.; Itokawa, H.; Takeya, K.; Kobayashi, J. i., "Samoquasine A, a Benzoquinazoline Alkaloid from the Seeds of Annona squamosa," J. Nat. Prod., 2002, 65, 1748-1748, DOI: 10.1021/np0204343.
(4) Yang, Y.-L.; Chang, F.-R.; Wu, Y.-C., "Total synthesis of 3,4-dihydrobenzo[h]quinazolin-4-one
and structure elucidation of perlolidine and samoquasine A," Tetrahedron Letters, 2003, 44, 319-322, DOI: 10.1016/S0040-4039(02)02577-7.
(5) Chakrabarty, M.; Sarkara, S.; Harigaya, Y., "An Expedient Synthesis of Benzo[h]quinazolin-4(3H)-one: Structure of Samoquasine A Revisited," Synthesis, 2003, 2292-2294, DOI: 10.1055/s-2003-42409.
InChIs
1: InChI=1/C12H8N2O/c15-12-10-6-5-8-3-1-2-4-9(8)11(10)13-7-14-12/h1-7H,(H,13,14,15)/f/h14H
InChIKey=BJVYARVTSUNBMW-YHMJCDSICO
2: InChI=1/C12H8N2O/c15-12-10-7-14-11-4-2-1-3-9(11)8(10)5-6-13-12/h1-7H,(H,13,15)/f/h13H
InChIKey=ULIAUQBOGQCMQM-NDKGDYFDCS
3: InChI=1/C12H8N2O/c15-12-10-6-5-8-3-1-2-4-9(8)11(10)7-13-14-12/h1-7H,(H,14,15)/f/h14H
InChIKey=JFJSVLSRVOIBPG-YHMJCDSICJ
4: InChI=1/C12H8N2O/c15-12-11-9(7-13-14-12)6-5-8-3-1-2-4-10(8)11/h1-7H,(H,14,15)/f/h14H
InChIKey=BGYAATPHYRJYHZ-YHMJCDSICO