A heterosubstituted corranulene analogue has now been prepared. Ito, Tokimaru, and Nozaki report the synthesis of 1 and compare it with corranulene.1 The x-ray structure of 1 shows it to be a deeper bowl than corranulene, and the bond distances suggest the Kekule structure with a central pyrrole and five Clar-type phenyl rings.
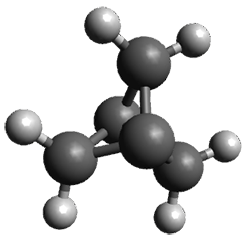
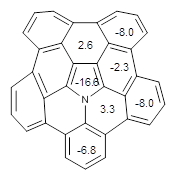
The B3LYP/6-311+G(2d,p) optimized structure of 2, then analogue of 1 missing the t-butyl group, is shown in Figure 1. Its geometry is very similar to that of 1 observed in the crystal structure. The NICS(0) values are shown in Scheme 1. These values support the notion of a central (aromatic) pyrrole surrounded by a periphery of five aromatic phenyl rings.
Scheme 1. NICS(0) values
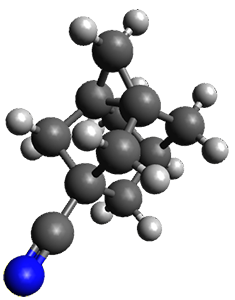
An interesting feature of bowl compounds is their inversion. The inversion barrier, through the planar TS shown in Figure 2, is computed to be 17.0 kcal mol-1 at B3LYP/6-311+G(2d,p). This is 6-7 kcal mol-1 larger than the inversion barrier of corranulene, which is not surprising given the additional phenyl groups about the periphery.
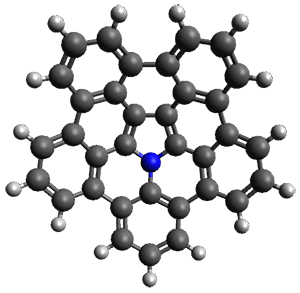
2 |
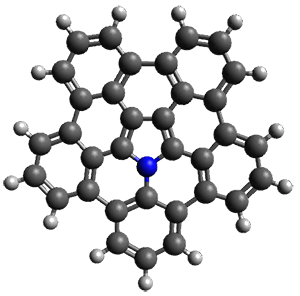
bowl inversion TS |
Figure 1. B3LYP/6-311+G(2d,p) optimized geometry of 2.
References
(1) Ito, S.; Tokimaru, Y.; Nozaki, K. "Benzene-Fused Azacorannulene Bearing an Internal Nitrogen Atom," Angew. Chem. Int. Ed. 2015, 54, 7256-7260, DOI: 10.1002/anie.201502599.
InChI
1: InChI=1S/C38H23N/c1-38(2,3)18-16-27-25-14-6-12-23-21-10-4-8-19-20-9-5-11-22-24-13-7-15-26-28(17-18)35(27)39-36(31(23)25)33(29(19)21)34(30(20)22)37(39)32(24)26/h4-17H,1-3H3
InChIKey= JJPREOOFFSVARV-UHFFFAOYSA-N
2: InChI=1S/C34H15N/c1-6-16-17-7-2-9-19-21-11-4-13-23-25-15-5-14-24-22-12-3-10-20-18(8-1)26(16)30-31(27(17)19)34(29(21)23)35(32(24)25)33(30)28(20)22/h1-15H
InChIKey= FFFUKUDWQMOFQQ-UHFFFAOYSA-N