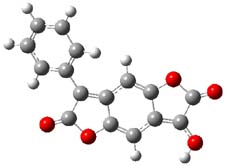
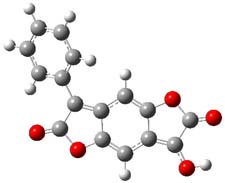
The keto-enol tautomerization is an interesting system for probing relative energies of subtle effects, playing off different bond type (and their associated strengths) with conjugation and hydrogen bonding and strain. Lawrence and Hutchings have now extended this to include the interplay of aromaticity and antiaromaticity in the keto-enol tautomerization of benzodifurantrione 1.1 The keto form 1k looks to be the favotable tautomer, containing an aromatic phenyl ring. The enol tautomer 1e requires the loss of that aromatic ring. Nonetheless, the enol structure is the only tautomer present in the crystal phase, and the enol tautomer is the dominant structure (if not the exclusive structure) in all solvents tested, including acetic acid, acetone, acetonitrile, chloroform, DMF, DMSO, propanol and toluene. The only solvents where the keto form is dominant are toluene and o-dichlorobenzene.
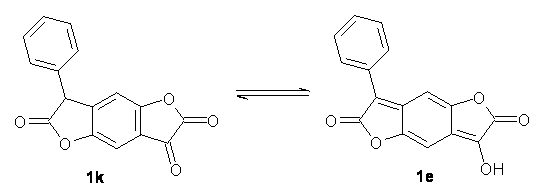
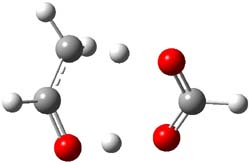
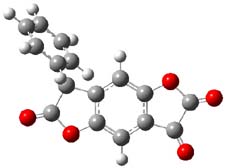
So, how does one rationalize this equilibrium? The B3LYP/6-311G(2d,p) structure of the two tautomers are shown in Figure 1. Note that there are two isomers of the enol form, differing on the orientation of the hydroxyl hydrogen. The syn isomer is the lowest energy form, in both the gas phase and in solution (PCM modeling acetonitrile, chlorobenzene and THF). So the enol form is the lowest energy structure when there are no special interactions involving hydrogen bonding or dipolar interactions with the solvent – there is an inherent energy preference for 1e.
Figure 1. B3LYP/6-311G(2d,p) structures of the tautomers of 1.1
To address that, they computed the NICS(0) values for each ring in the two tautomers. The pendant phenyl group is aromatic in both structures, as expected. The lactone ring has NICS values near 0 in both structures. The interior phenyl ring is aromatic (NICS = -7.5) in 1k but is non-aromatic in 1e, with NICS=-0.4. So the aromaticity of this ring is lost upon enolization, and thus would favor 1k. However, the terminal ring in the keto tautomer has NICS = +7.2, suggesting that it is antiaromatic, and upon enolization, the ring becomes slightly aromatic, with NICS = -2.1. Thus, the keto form is plagued by an antiaromatic ring, which is then lost in the enol form. The result is the interplay between losing an aromatic ring and its stabilization when the enol is formed balanced by also losing an antiaromatic ring with its destabilization. The authors do not offer any quantization (rightfully so!) of the stabilization/destabilization associated with these rings. But very subtle effects are clearly at play.
References
(1) Lawrence, A. J.; Hutchings, M. G.; Kennedy, A. R.; McDouall, J. J. W., "Benzodifurantrione: A Stable Phenylogous Enol," J. Org. Chem., 2010, 75, 690–701, DOI: 10.1021/jo9022155
InChIs
1k: InChI=1/C16H8O5/c17-14-10-7-11-9(6-12(10)21-16(14)19)13(15(18)20-11)8-4-2-1-3-5-8/h1-7,13H
InChIKey=GNWKSKHSRUSFBC-UHFFFAOYAC
1e: InChI=1/C16H8O5/c17-14-10-7-11-9(6-12(10)21-16(14)19)13(15(18)20-11)8-4-2-1-3-5-8/h1-7,17H
InChIKey=MZLQKOSFMRKQIO-UHFFFAOYAB