Optical activity is a major tool for identifying enantiomers. With recent developments in computational techniques, it is hoped that experiments combined with computations will be a powerful tool for determining absolute configuration. The recent work of Lahiri, et al. demonstrates the scope of theoretical work that is still needed to really make this approach broadly applicable.1
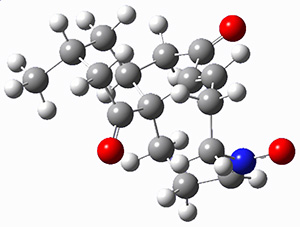
They prepared (1R,4R)-norbornenone 1 and measured its optical rotation in the gas phase and in dilute solutions of acetonitrile and cyclohexane. The specific rotations at three different wavelengths are listed in Table 1. Of first note is that though there is some small differences in solution, as expected, there really is substantial differences between the gas- and solution phases. Thus cautionary point 1: be very careful of comparing solution phase experimental optical activity with computed gas phase predictions.
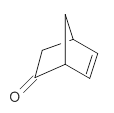
1
Table 1. Experimental and computed specific rotation of 1.
|
|
355.0 nm |
589.3 nm |
633.0 nm |
|
Gas phase |
|||
|
Expt |
6310 |
755 |
617 |
|
B3LYP |
10887 |
1159 |
944 |
|
CCSD |
3716 |
550 |
453 |
|
Acetonitrile solution |
|||
|
Expt |
8607 |
950 |
776 |
|
PCM/B3LYP |
11742 |
1277 |
1040 |
|
Cyclohexane solution |
|||
|
Expt |
9159 |
981 |
799 |
|
PCM/B3LYP |
11953 |
1311 |
1069 |
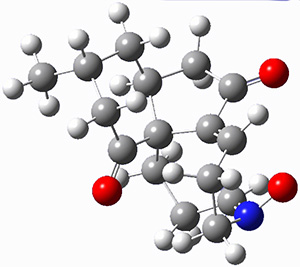
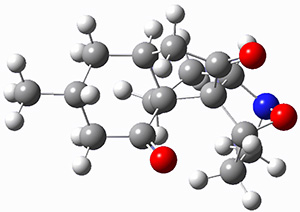
For the computations, the geometry of 1 was optimized at B3LYP/aug-cc-pVTZ (see Figure 1. The OR was computed at B3LYP with different basis sets, finding that the difference in the predicted specific rotation at 598.3nm differs only quite little (90.6 deg dm-1 (g/mL)-1) between the computations using aug-cc-pVTZ and aug-cc-pVQZ) suggesting that the basis set limit has been reached. The gas –phase computed values at B3LYP and CCSD are also shown in Table 1. Though these computations do get the correct sign of the rotation, they are appreciably off of the experimental values, and the percent error varies with wavelength. Cautionary point 2: it is not obvious what is the appropriate computational method to compute OR, and beware of values that seem reasonable at one wavelength – this does not predict a good agreement at a different wavelength.
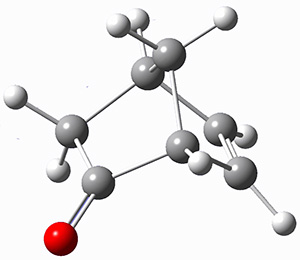 |
Figure 1. Optimized geometry of 1 at B3LYP/aug-cc-pVTZ.
Lastly, computed solution values of the OR were performed with PCM and B3LYP. These values are given in Table 1. Again the agreement is poor. So cautionary point 3: computed (PCM) solution OR
may be in quite poor agreement with experiment.
Often the culprit to poor agreement between computed and experimental OR is attributed to omitted vibrational effects, but in this case, because 1 is so rigid, one might not expect too much error to be caused by the effects of vibrations. So the overall result – we need considerable work towards addressing how to accurately compute optical activity!
References
(1) Lahiri, P.; Wiberg, K. B.; Vaccaro, P. H.; Caricato, M.; Crawford, T. D. "Large Solvation Effect in the Optical Rotatory Dispersion of Norbornenone," Angew. Chem. Int. Ed. 2014, 53, 1386-1389, DOI: 10.1002/anie.201306339.
InChIs
1: InChI=1S/C7H8O/c8-7-4-5-1-2-6(7)3-5/h1-2,5-6H,3-4H2/t5-,6+/m1/s1
InChIKey=HUQXEIFQYCVOPD-RITPCOANSA-N