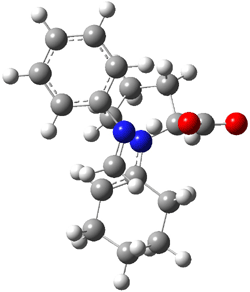
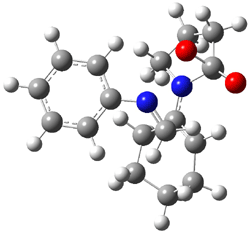
Here are two nice examples of the use of computed spectra in identifying the structure of large molecules.
Castle and co-workers describe the synthesis of what they hoped would be runanine 1.1 However, after they had completed their synthesis, the 1H NMR spectrum of their product differed significantly from that of runanine. Further the optical rotation of 1 is -400, while that of their product is -34. Speculating on what might be the product they came up with 4 alternative structures 2-5. The 13C NMR of 1-5 were then computed by optimizing the structures at mPW1PW91/6-31G* followed by a GIAO computation at mPW1PW91/aug-cc-pVDZ with PCM (solvent is chloroform). The differences between the computed chemical shifts for 1-5 and the experimental shifts of the obtained product are summarized in Table 1. The authors conclude that their product is 5, a compound they name isorunanine.
|
|
|
|
|
|
|
|
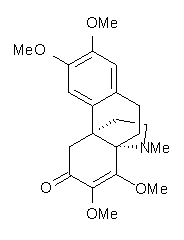
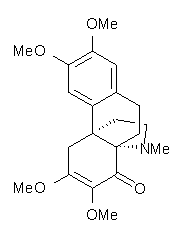
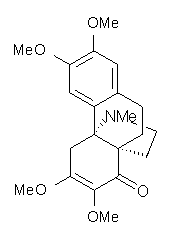
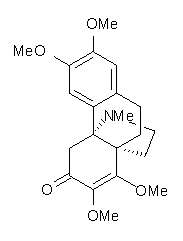
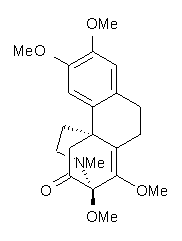
Table 1. Average difference and maximum difference between the computed and experimental 13 C chemical shifts (ppm).
|
Compound |
Average Δδ |
Maximum Δδ |
|
1 |
9.7 |
61.7 |
|
2 |
9.3 |
60.1 |
|
3 |
12.0 |
54.6 |
|
4 |
10.9 |
61.8 |
|
5 |
3.8 |
9.8 |
The authors also report the rather poor agreement between the computer spectrum of 6 and the experimental spectrum in benzene. Unfortunately, not enough details are provided to really determine where errors might be occurring. For example, there is no indication of examining multiple conformations (and those methoxy groups can rotate along with the inversion at the amine). Once again, the supporting materials, while extensive in terms of experimental NMR spectra, contain no details of the computed structures.
The structure of hypurticin 6 was determined using a comparison of computed coupling constants.2 Here the authors first assumed that four possible stereoisomers are possible 6a-d, given that the other stereocenters were determined unambiguously by experiment and biogenesis considerations. B3LYP/6-31G(d) optimization of a restricted set of conformations led to the lowest energy conformer. The coupling constants computed for these four structures indicated the closet agreement between the computed constants of 6a with experimental values. An exhaustive search of the conformational space of each of these diastereomers at B3LYP/DGDZVP followed by Boltzmann weighting of the coupling constants confirmed that 6a is the structure of hypurticin.

References
(1) Nielsen, D. K.; Nielsen, L. L.; Jones, S. B.; Toll, L.; Asplund, M. C.; Castle, S. L., "Synthesis of Isohasubanan Alkaloids via Enantioselective Ketone Allylation and Discovery of an Unexpected Rearrangement," J. Org. Chem. 2009, 74, 1187-1199, DOI: 10.1021/jo802370v.
(2) Mendoza-Espinoza, J. A.; Lopez-Vallejo, F.; Fragoso-Serrano, M.; Pereda-Miranda, R.; Cerda-Garcia-Rojas, C. M., "Structural Reassignment, Absolute Configuration, and Conformation of Hypurticin, a Highly Flexible Polyacyloxy-6-heptenyl-5,6-dihydro-2H-pyran-2-one," J. Nat. Prod. 2009, 72, 700-708, DOI: 10.1021/np800447k.