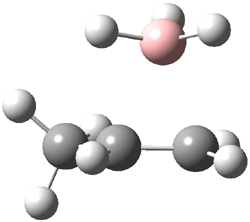
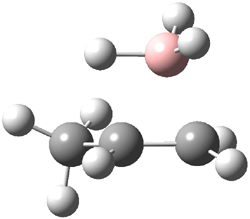
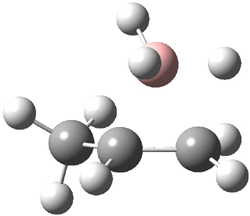
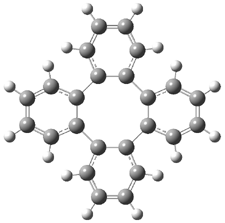
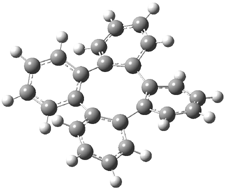
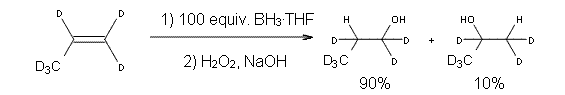Careful consideration of orbital topolologies of pericyclic reactions has led to the recent flurry of activity related to Möbius aromaticity, homoaromaticity, and antiaromaticity. I discussed this briefly in Chapter 2 of the book and in these posts (1, 2, 3). Mauksh and Tsogoeva1 have clearly demoted the four different topologies of the transition state of pericyclic reactions. One need to be concerned about (a) the topology of the molecule (does it will have the familiar twist of a Möbius strip or not?) and (b) the topology of the π-system (is there a phase inversion or not?). These four topologies are shown in Figure 1. The pink stick represents the positive lobe of the carbon p orbital.
 (A) |
 (B) |
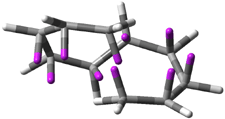 (C) |
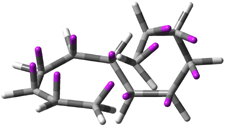 (D) |
Figure 1. Topologies of electrocyclization reactions.
Three of these possibilities had been previously identified. The first (Fig. 1A) is the TS for the electrocyclization of 1,3,5-hexadiene. It has both Hückel topology of the molecule and the p orbitals. The second example (Fig. 1B) is the classic Zimmerman example of the electrocyclization of all-cis 1,3,5,7-octatetraene. It has Hückel topology of the molecule but one phase inversion of the p orbitals. The third example (Fig 1C) is the electrocyclization of (3E,5Z,7E)-1,3,5,7,9-decapentaene, proposed by Rzepa.2 Here we have a Möbius topology but there is no phase inversion of the p orbitals.
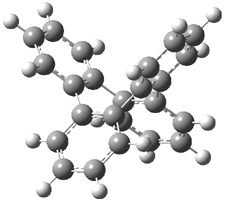
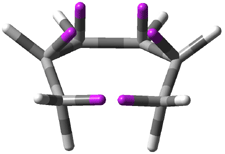
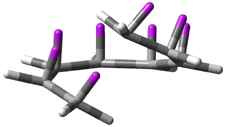
Mauksch and Tsogoeva report on the novel electrocylization of (3E,5E,7E,9E)-1,3,5,7,9,11-dodecahexaene (1), the fourth topology type (Fig 1d).1 Here the molecule has the Möbius topology and there is one phase inversion. Figure 2 displays the geometries of the reactant 1, the electrocyclization transition state 2, and the product 3. The activation barrier is 35.7 kcal mol-1. The NICS value at the center of the ring of the transition state is -12.8ppm , indicative of aromatic character, which is supported by the very small variation of the C-C distances (less than 0.02 Å).
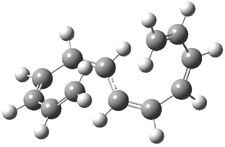 1 |
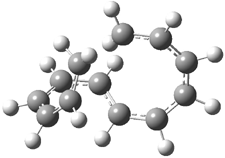 2 |
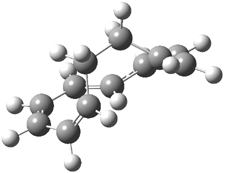 3 |
Figure 2. B3LYP/6-31G* optimized geometries of 1-3.1
Henry Rzepa has commented on this paper in his blog, along with detailing another example of this type of topology. In a second post, Henry discusses the issue of competition between aromatic and antiaromatic character in a related molecule.
References
(1) Mauksch, M.; Tsogoeva, S. B., "A Preferred Disrotatory 4n Electron Möbius Aromatic Transition State for a Thermal Electrocyclic Reaction," Angew. Chem. Int. Ed., 2009, 48, 2959-2963, DOI: 10.1002/anie.200806009
(2) Rzepa, H. S., "Double-twist Möbius aromaticity in a 4n+2 electron electrocyclic reaction," Chem. Commun., 2005, 5220-5222, DOI: 10.1039/b510508k.
InChIs
(3Z)-1,3,5-hexatriene: InChI=1/C6H8/c1-3-5-6-4-2/h3-6H,1-2H2/b6-5-
InChIKey=AFVDZBIIBXWASR-WAYWQWQTBR
(3Z,5Z)-octa-1,3,5,7-tetraene: InChI=1/C8H10/c1-3-5-7-8-6-4-2/h3-8H,1-2H2/b7-5-,8-6-
InChIKey=VXQUABLSXKFKLO-SFECMWDFBK
(3Z,5E,7Z)- 1,3,5,7,9-decapentaene: InChI=1/C10H12/c1-3-5-7-9-10-8-6-4-2/h3-10H,1-2H2/b7-5-,8-6-,10-9+
InChIKey=XKWRJEBRBGIQBA-LODOGNSSBI
(3Z,5Z,7Z,9Z)- 1,3,5,7,9,11-dodecahexaene (1): InChI=1/C12H14/c1-3-5-7-9-11-12-10-8-6-4-2/h3-12H,1-2H2/b7-5-,8-6-,11-9-,12-10-
InChIKey=GLCGEJFIZRSXBL-CAVFYFSLBM
(1Z,3Z,5E,7Z,9Z)-cyclododeca-1,3,5,7,9-pentaene (3): InChI=1/C12H14/c1-2-4-6-8-10-12-11-9-7-5-3-1/h1-10H,11-12H2/b2-1+,5-3-,6-4-,9-7-,10-8-
InChIKey=DULJMQFBRDFTQN-UWFJMCQMBK