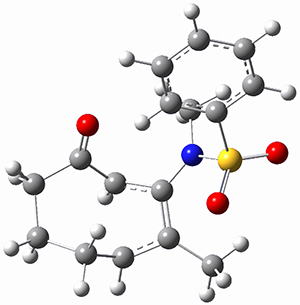
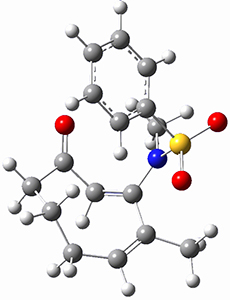
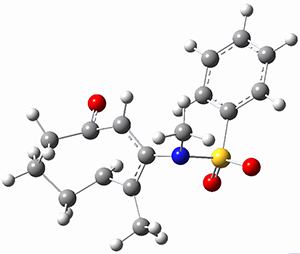
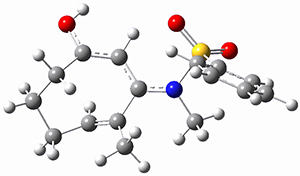
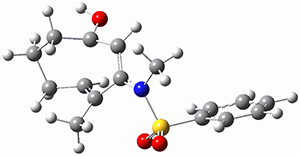
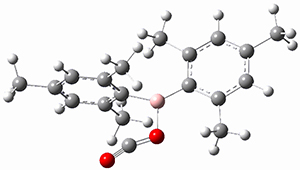
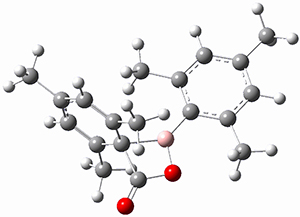
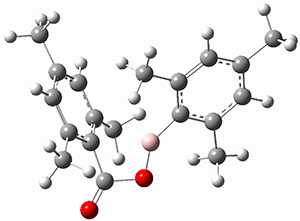
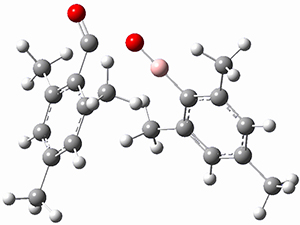
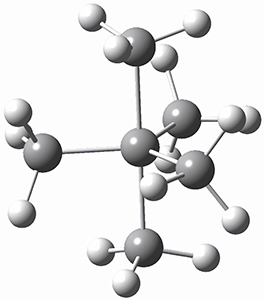
Trying to get carbon to bond in unnatural ways seems to be a passion for many organic chemists! Schleyer has been interested in unusual carbon structures for decades and he and Schaefer now report a molecule with a pentacoordinate carbon bound to five other carbon atoms. Their proposed target is pentamethylmethane cation C(CH3)5+ 1.1 The optimized geometry of 1, which has C3h symmetry, at MP2/cc-pVTZ is shown in Figure 1. The bonds from the central carbon to the equatorial carbon are a rather long 1.612 Å, but the bonds to the axial carbon are even longer, namely 1.736 Å. Bader analysis shows five bond critical points, each connecting the central carbon to one of the methyl carbons. Wiberg bond index and MO analysis suggests that the central carbon is tetravalent, with a 2-electron-3-center bond involving the central and axial carbons.
1 |
|
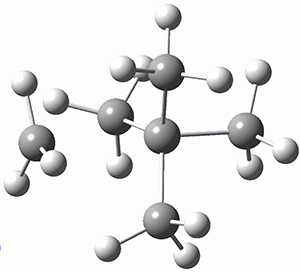
TS1 |
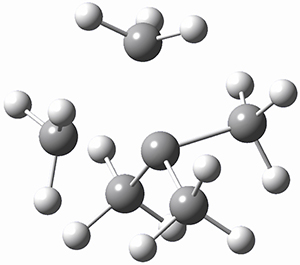
TS2 |
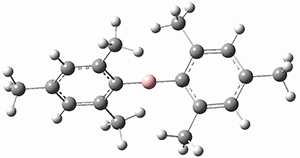
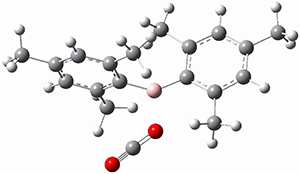
Figure 1. MP2/cc-pVTZ optimized geometries of 1 and dissociation transition states.
So while 1 is a local energy minimum, it sits in a very shallow well. One computed dissociation path, which passes through TS1 (Figure 1) on its way to 2-methyl-butyl cation and methane has a barrier of only 1.65 kcal mol-1 (CCSD(T)/CBS + ZPE). A second dissociation pathway goes through TS2 to t-butyl cation and ethane with a barrier of only 1.34 kcal mol-1. Worse still is that the free energy estimates suggest “spontaneous dissociation … through both pathways”.
Undoubtedly, this will not be the last word on trying to torture a poor carbon atom.
References
(1) McKee, W. C.; Agarwal, J.; Schaefer, H. F.; Schleyer, P. v. R. "Covalent Hypercoordination: Can Carbon Bind Five Methyl Ligands?," Angew. Chem. Int. Ed. 2014, 53, 7875-7878, DOI: 10.1002/anie.201403314.
InChIs
1: InChI=1S/C6H15/c1-6(2,3,4)5/h1-5H3/q+1
InChIKey=GGCBGJZCTGZYFV-UHFFFAOYSA-N