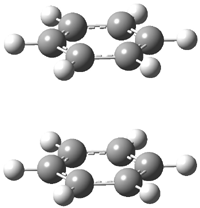
The nature of the bridgehead-bridgehead bond in [1.1.1]propellane 1 poses an interesting quandary. The bond involves two inverted carbon atoms, whose hybrids should point away from each other. The internuclear region has in fact much less electron density than for an ordinary C-C bond. Nonetheless, the molecule is stable and the C-C bond is estimated to have a strength of about 6 kcal mol-1.

Shaik and Hiberty1 have now proposed that the central C-C bond of [1.1.1]propellane is a charge-shift bond. In classical valence bond theory, we have three configurations for a bond: the covalent structure A↑↓B ↔ A↓↑B, and the two ionic structures A↑↓ B and A B↑↓. The description of a typical covalent bond is dominated by the covalent VB structure with a little bit of the ionic structures mixed in. A charge-shift bond is one where the resonance energy due to the mixing of the covalent and ionic structures mostly accounts for the stabilization of the bond.2 Just such a case is found in the F-F bond, and also to for the central C-C bond of [1.1.1]propellane!
References
(1) Wu, W.; Gu, J.; Song, J.; Shaik, S.; Hiberty, P. C., "The Inverted Bond in [1.1.1]Propellane is a Charge-Shift Bond," Angew. Chem. Int. Ed., 2008, ASAP DOI: 10.1002/anie.200804965
(2) Shaik, S.; Danovich, D.; Silvi, B.; Lauvergnat, D. L.; Hiberty, P. C., "Charge-Shift Bonding – A Class of Electron-Pair Bonds That Emerges from Valence Bond Theory and Is Supported by the Electron Localization Function Approach," Chem. Eur. J., 2005, 11, 6358-6371, DOI: 10.1002/chem.200500265
InChI
1: InChI=1/C5H6/c1-4-2-5(1,4)3-4/h1-3H2
InChIKey=ZTXSPLGEGCABFL-UHFFFAOYAJ