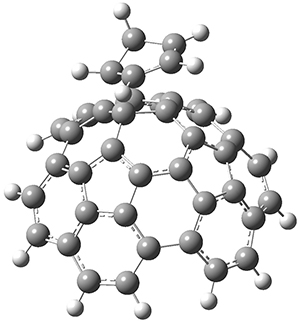
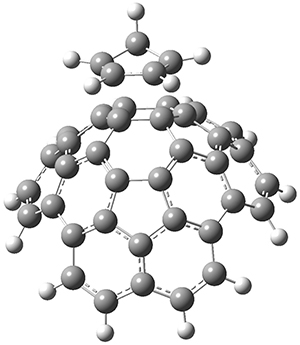
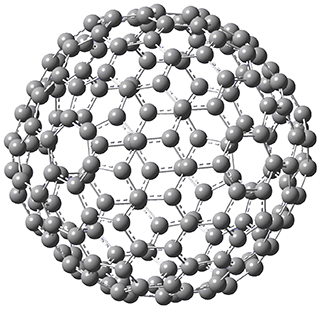
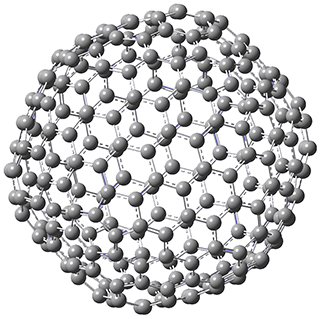
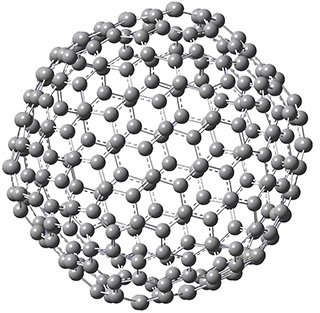
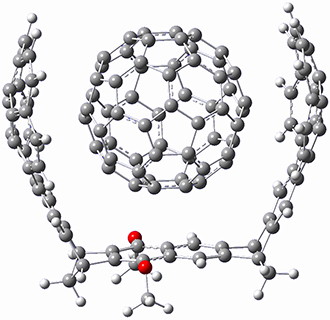
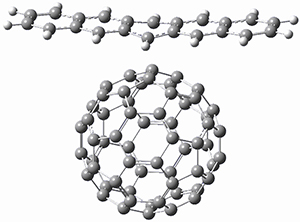
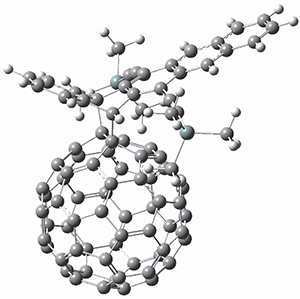
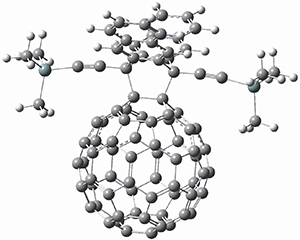
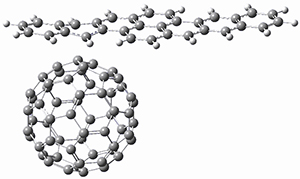
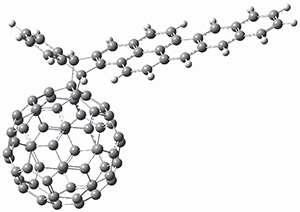
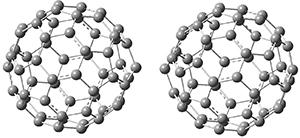
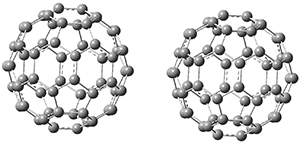
The Grimme group has examined all 1812 C60 isomers, in part to benchmark some computational methods.1 They computed all of these structures at PW6B95-D3/def2-QZVP//PBE-D3/def2-TZVP. The lowest energy structure is the expected fullerene 1 and the highest energy structure is the nanorod 2 (see Figure 1).
|
|
|
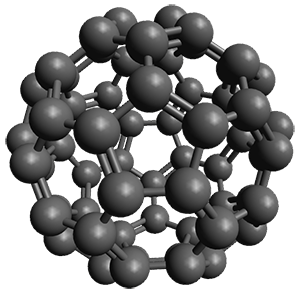
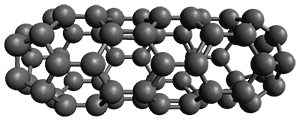
Figure 1. Optimized structures of the lowest (1) and highest (2) energy C60 isomers.
About 70% of the isomers like in the range of 150-250 kcal mol-1 above the fullerene 1, and the highest energy isomer 2 lies 549.1 kcal mol-1 above 1. To benchmark some computational methods, they selected the five lowest energy isomers and five other isomers with higher energy to serve as a new database (C60ISO), with energies computed at DLPNO-CCSD(T)/CBS*. The mean absolute deviation of the PBE-D3/def2-TZVP relative energies with the DLPNO-CCSD(T)/CBS* energies is relative large 10.7 kcal mol-1. However, the PW6B95-D3/def2-QZVP//PBE-D3/def2-TZVP method is considerably better, with a MAD of only 1.7 kcal mol-1. This is clearly a reasonable compromise method for fullerene-like systems, balancing accuracy with computational time.
They also compared the relative energies of all 1812 isomers computed at PW6B95-D3/def2-QZVP//PBE-D3/def2-TZVP with a number of semi-empirical methods. The best results are with the DFTB-D3 method, with an MAD of 5.3 kcal mol-1.
References
1) Sure, R.; Hansen, A.; Schwerdtfeger, P.; Grimme, S., "Comprehensive theoretical study of all 1812 C60 isomers." Phys. Chem. Chem. Phys. 2017, 19, 14296-14305, DOI: 10.1039/C7CP00735C.
InChIs
1: InChI=1S/C60/c1-2-5-6-3(1)8-12-10-4(1)9-11-7(2)17-21-13(5)23-24-14(6)22-18(8)28-20(12)30-26-16(10)15(9)25-29-19(11)27(17)37-41-31(21)33(23)43-44-34(24)32(22)42-38(28)48-40(30)46-36(26)35(25)45-39(29)47(37)55-49(41)51(43)57-52(44)50(42)56(48)59-54(46)53(45)58(55)60(57)59
InChIKey=XMWRBQBLMFGWIX-UHFFFAOYSA-N
2: InChI=1S/C60/c1-11-12-2-21(1)31-41-32-22(1)3-13(11)15-5-24(3)34-43(32)53-55-47-36-26-6-16-17-7(26)28-9-19(17)20-10-29-8(18(16)20)27(6)37-46(36)54(51(41)55)52-42(31)33-23(2)4(14(12)15)25(5)35-44(33)58-56(52)48(37)39(29)50-40(30(9)10)49(38(28)47)57(53)59(45(34)35)60(50)58
InChIKey=AGZHNPDQKMDYHI-UHFFFAOYSA-N