What happens when anions are sequentially solvated with more and more water? In an interesting study by Chesnovsky, Gerber and coworkers, the answer is proton transfer!1 They examined the conjugate base of aniline (C6H5NH–, 1) using both PES and MP2/DZP computations. The PES spectrum of 1 shows two strong peaks. When this anion is then coordinates with one or two water molecules, C6H5NH–).(H2O) or (C6H5NH–).(H2O)2, the peaks shifts to higher energy but the general shape remains unchanged. When three or more water molecules are coordinated to 1, the PE spectra totally changes, becoming broad and relatively featureless.
What accounts for this different PES? The authors posit that the PES of these larger clusters resembles that of hydrated OH– clusters. Optimization of (C6H5NH–).(H2O) or (C6H5NH–).(H2O)2
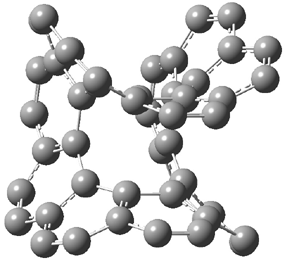
gave structures of the waters hydrogen bonded to the nitrogen anion. However, optimization of the (C6H5NH–).(H2O)3 gave in fact a cluster of the form (C6H5NH2).(HO–).(H2O)2. The two lowest energy structures are shown in Figure 1. The structures correspond to the transfer of one proton from water to the anilinide anion to give aniline associated with hydroxide and two water molecules.
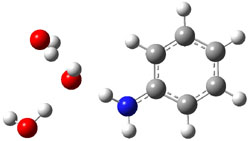 Rel E: 0.0 |
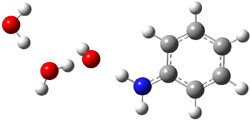 Rel E: 0.10 eV |
Figure 1. B3LYP/6-31+G(d) optimized structures of
the C6H5NH2).(HO–).(H2O)2 clusters.
(Note: Once again the authors have failed to include the computed structures as part of the supporting information and so I have reoptimized the structures but at a lower, computationally more tractable level. Hopefully, authors, editors and reviewers will standardize this practice and include such materials in all papers in the very near future!)
Though aniline is a stronger gas-phase acid than water (DPE(aniline) = 366.4 kcal mol-1 and DPE(H2O) = 390.3 kcal mol-1), the reverse is true in solution (pKa(aniline) = 27.3) and pKa(water) = 15.7). As more water molecules are present, the preferential solvation of the hydroxide anion over C6H5NH– results in the formation of hydroxide.
References
(1) Wolf, I.; Shapira, A.; Giniger, R.; Miller, Y.; Gerber, R. B.; Cheshnovsky, O., "Critical Size for Intracluster Proton Transfer from Water to an Anion," Angew. Chem. Int. Ed., 2008, DOI: 10.1002/anie.200800542