Host-guest recognition is a major theme of modern chemistry. Computation of these systems remains a real challenge for many reasons, especially the typically large size of the molecules involved and the need for accurately computing weak, non-covalent interactions. This latter point remains a major problem with density functional theory.
Goddard has now examined a rotaxane system.1 Goddard employed a variety of functionals (B3LYP. PBE, and MO6 variants) to 1, a compound prepared by Stoddart.2 The counterion of the experimentally prepared rotoxane is PF6–; in the computations, Goddrad employed either no counterions or four chloride ions.
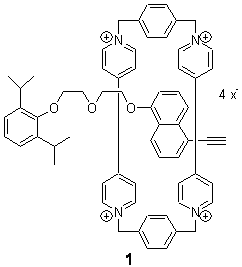
The optimized structure of 1 without counterions computed at B3LYP/6-31G** and MO6-L/6-31G** are shown in Figure 1. The major difference in these structures is the orientation of the naphthyl group inside the host. B3LYP predicts that it is skewed, while MO6-L predicts that it lies parallel to the bipyridinium side. The x-ray structure has the parallel structure, similar to that found with MO6-L, though the pendant bis-i-proylphenyl ring is farther down in the x-ray structure than in the computed structure.
|
(a) 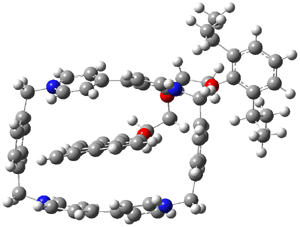 |
(b) 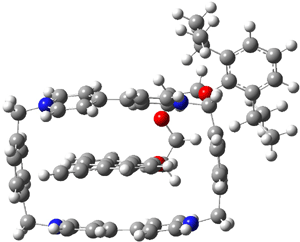 |
Figure 1. Optimized structure of 14+
(a) B3LYP/6-31G** and (b) MO6-L/6-31G**.1
None of the methods perform particularly well in computing the binding energy of the host and guest. The experimental value is -4.9 ± 1 kcal mol-1. In the gas phase, the two methods predict that the system is bound, -24.9 (B3LYP, -75.2 kcal mol-1, MO6-L). In acetonitrile, B3LYP predicts that it is unbound, while MO6-L predicts a binding energy of -27.5 kcal mol-1. Inclusion of four chloride ions leads to some improvement in the binding energy in the gas phase but not for the solution phase.
The excitation energy is 3.50 eV.Computation of the excitation energy is poor with B3LYP (1.33 eV) but nearly exact with MO6-HF//MO6-L (3.42 eV).
Goddard concludes that computation of these sort of interlocked molecules should be performed with the MO6 family of functionals, but clearly more work is needed if accurate energies are required.
References
(1) Benitez, D.; Tkatchouk, E.; Yoon, I.; Stoddart, J. F.; Goddard, W. A., "Experimentally-Based Recommendations of Density Functionals for Predicting Properties in Mechanically Interlocked Molecules," J. Am. Chem. Soc., 2008, 130, 14928-14929, DOI: http://dx.doi.org/10.1021/ja805953u.
(2) Nygaard, S.; Leung, K. C. F.; Aprahamian, I.; Ikeda, T.; Saha, S.; Laursen, B. W.; Kim, S.-Y.; Hansen, S. W.; Stein, P. C.; Flood, A. H.; Stoddart, J. F.; Jeppesen, J. O., "Functionally Rigid Bistable [2]Rotaxanes," J. Am. Chem. Soc., 2007, 129, 960-970, DOI: http://dx.doi.org/10.1021/ja0663529
InChIs
Guest: InChI=1/C28H32O3/c1-6-22-10-7-14-26-25(22)13-9-15-27(26)30-18-16-29-17-19-31-28-23(20(2)3)11-8-12-24(28)21(4)5/h1,7-15,20-21H,16-19H2,2-5H3
InChIKey=HYRADSXCSHFXAZ-UHFFFAOYAJ
Host: InChI=1/C36H32N4/c1-2-30-4-3-29(1)25-37-17-9-33(10-18-37)35-13-21-39(22-14-35)27-31-5-7-32(8-6-31)28-40-23-15-36(16-24-40)34-11-19-38(26-30)20-12-34/h1-24H,25-28H2/q+4
InChIKey=URORLZXVTFVIPS-UHFFFAOYAV

Computational Organic Chemistry » Origin of DFT failure responded on 19 May 2009 at 8:08 am #
[…] with some seemingly straightforward reactions (as discussed in these previous blog posts: A, B, C, D, E, F) has become a bit clearer. Brittain and co-workers have identified the culprit.1 They […]