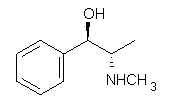
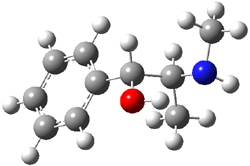
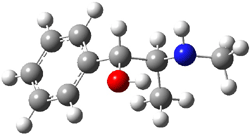
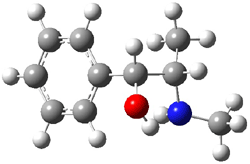
Yet more on the benzene dimer. Lesczynski has optimized 9 different benzene dimer configurations, shown in Scheme 1.1 There are two T-shaped isomers, where a hydrogen from one benzene interacts with the center of the π-cloud of the second. There are two bent versions of the T-shape, called Bent-T-shape. There are two sandwich configurations and two variants where the benzenes are parallel but displaced. Lastly, they report on a new variant, the V-shape configuration. (Once again, the author has not deposited the structures and so I can’t produce interactive figures!)
Scheme 1
|
|
|
|
|
|
|
|
|
|
|
|
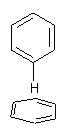
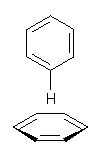
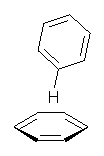
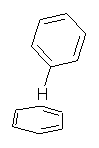
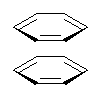
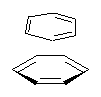
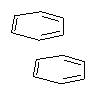
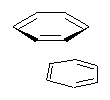
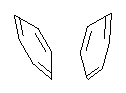
The structures were optimized at MP2/aug-cc-pVDZ and then single point energies computed at MP4(SDTQ)/aug-cc-pVDZ and corrected for basis set superposition error. I list these energies in Table 1. They authors note that in comparison with CCSD(T) computations one has to adjust the amount of BSSE correction – which just supports my long-held contention that the standard counterpoise correction overcompensates and that we really have no reliable way of correcting for BSSE.
Table 1. Dimerization energies (kcal mol-1) at MP4(SDTQ)/aug-cc-pVDZ.1
|
T-1 |
T-2 |
BT-1 |
BT-2 |
SW-1 |
SW-2 |
PD-1 |
PD-2 |
V |
The relative energies of the 9 configurations are similar, indicating a very flat potential energy surface. The lowest energy structure is BT-2, and the V-shape configuration is the least favorable of the nine geometries examined.
References
(1) Dinadayalane, T. C.; Leszczynski, J., "Geometries and stabilities of various configurations of benzene dimer: details of novel V-shaped structure revealed " Struct. Chem. 2009, 20, 11-20, DOI: 10.1007/s11224-009-9411-6.