Alonso and coworkers have developed the technique of laser ablation molecular beam Fourier transform microwave spectroscopy to detect biomolecules. In a recent paper1 they determined the structure of the glycine:one water complex – it is of the neutral configuration. They have now examined the conformations of cysteine2. The presence of the thiol side group adds considerable complexity to the problem due to the many conformations possible.
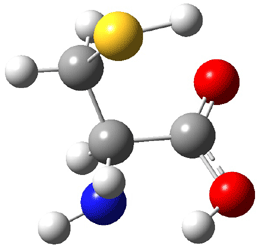
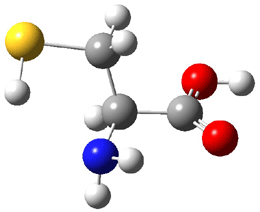
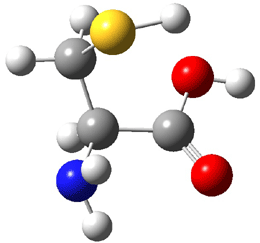
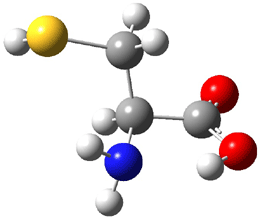
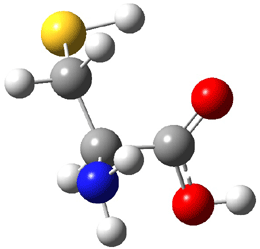
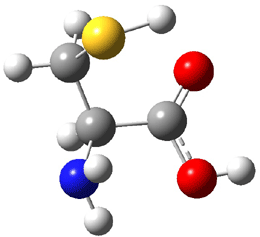
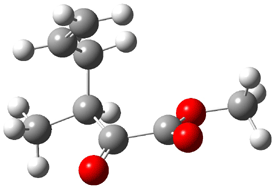
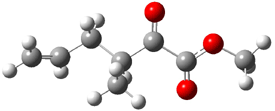
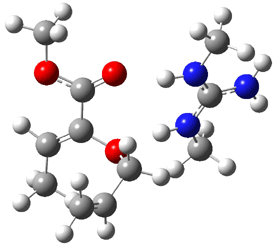
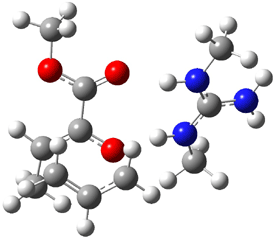
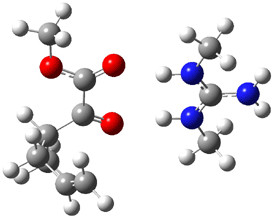
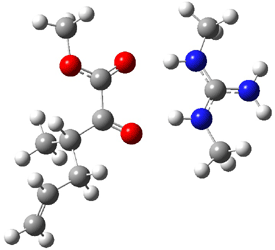
The experiment detected six conformers. Determining the structures responsible for each set of signals was made possible by comparing the experimental results with those determined by computation. Alonso computed 11 low energy conformations of cysteine at MP2/6-311++G(d,p). Then comparing the computed rotational constants and 14N nuclear quadrupole coupling tensor components with the experiment, they were able to match up all six experimental conformers with computed structures. The experimental and computed constants for the three most abundant structures are listed in Table 1. The geometries of all six conformers are drawn in Figure 1.
Table 1.Experimental and computed spectroscopic constants (MHz) for the three most abundant conformers of cysteine.2
|
|
IIb
|
Ia
|
Ib
|
|
|
Expt
|
MP2
|
Expt
|
MP2
|
Expt
|
MP2
|
|
A
|
3071.14
|
3040
|
4235.63
|
4221
|
2889.45
|
2855
|
|
B
|
1606.54
|
1623
|
1187.28
|
1185
|
1623.00
|
1664
|
|
C
|
1331.80
|
1347
|
1003.11
|
1013
|
1367.83
|
1386
|
|
χaa
|
-3.12
|
-3.14
|
-4.26
|
-4.67
|
-0.14
|
-0.01
|
|
χbb
|
2.44
|
2.59
|
2.78
|
2.86
|
0.44
|
0.25
|
|
χcc
|
0.68
|
0.55
|
1.49
|
1.80
|
-0.30
|
-0.24
|
|
ΔEa
|
|
0
|
|
450
|
|
325
|
aRelative energy in cm-1 computed at MP4/6-311++G(d,p)// MP2/6-311++G(d,p).
|
IIb (0.0)
|
Ia (450)
|
|
Ib (325)
|
IIa (527)
|
|
IIIβc (765)
|
IIIβb (585)
|
Table 1. Optimized structures of the six observed conformers of cysteine. Relative energies in cm-1 computed at MP4/6-311++G(d,p)//MP2/6-311++G(d,p). (Note – the geometries shown were optimized at PBE1PBE/6-311+G(d,p) since they MP2 structures are not available!)
This study demonstrates the nice complementary manner in which computation and experiment can work together in structure determination.
References
(1) Alonso, J. L.; Cocinero, E. J.; Lesarri, A.; Sanz, M. E.; López, J. C., "The Glycine-Water
Complex," Angew. Chem. Int. Ed. 2006, 45, 3471-3474, DOI: 10.1002/anie.200600342
(2) Sanz, M. E.; Blanco, S.; López, J. C.; Alonso, J. L., "Rotational Probes of Six Conformers of Neutral Cysteine," Angew. Chem. Int. Ed. 2008, DOI: 10.1002/anie.200801337
InChI
Cysteine: InChI=1/C3H7NO2S/c4-2(1-7)3(5)6/h2,7H,1,4H2,(H,5,6)/t2-/m0/s1
InChIKey: XUJNEKJLAYXESH-REOHCLBHBU