Li and Danishefsky report a study of the Diels-Alder reaction involving cyclobutenone 1 as the dienophile.1 They claim that “perhaps the ring strain of 1 might well serve to enhance its dienophilicity relative to corresponding cyclopentenones or cyclohexenones.” In fact, 1 is an excellent dienophile, with reactions at or below 0° being accomplished in less than half a day with yields upwards of 90%. The reaction goes with endo selectivity.
What is surprising to me is the statement in the article:
While the magnitude of the effect could not have been predicted in advance, the rate enhancement with 1 must reflect the favorable effects of rehybridization of two particularly strained sp2 carbons in the cycloaddition transition state.
Now, Danishefsky alludes to upcoming computations results in a future paper, but I don’t see why the rate enhancement could not have been “predicted in advance”. So, I have optimized the structures of reactants, endo and exo transition states, and products of the reaction of 1,3-butadiene with 1, cyclopentenone 2 and cyclohexenone 3 at B3LYP/6-311G(d) – Reactions 1-3.
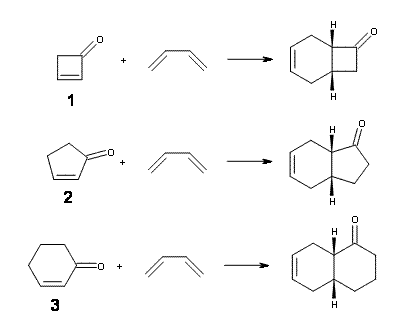
The endo TS is preferred for the reaction of 1 and 2, while the endo and exo TSs for 3 are essentially isoenergetic. The optimized geometries are shown in Figure 1.
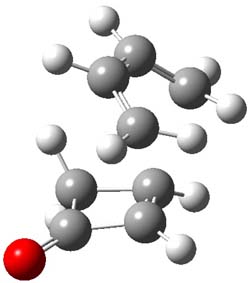 1TSendo |
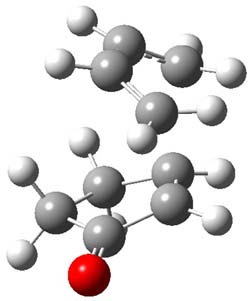 2TSendo |
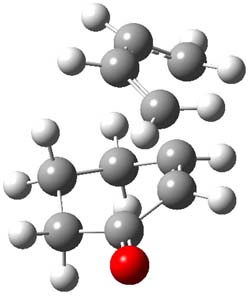 3TSendo |
Figure 1. B3LYP/6-311G(d) optimized geometries of the endo TSs of Reactions 1-3.
The computed activation barriers and overall reaction energies are listed in Table 1. Clearly, the cycloaddition of 1 is favored both in terms of kinetics (having the lowest barrier) and thermodynamically (having the most exothermic reaction energy). In fact, the reaction barriers increases in going from 1 to 2 to 3 and the exothermicity decreases in that same order. This nicely dovetails with the strain energies of the dienophiles and the fact that cyclopententones and cyclohexenones are generally poor dienophiles. Thus, one clearly could have predicted these results in advance!
Table 1. Activation and Reaction Energy (kcal mol-1) for Reactions 1-3.
|
Reaction |
Ea |
ΔE |
|
1 |
18.8 |
-35.2 |
|
2 |
24.1 |
-27.1 |
|
3 |
25.7 |
-27.1 |
Nonetheless, the experimental work is extremely nice and this work offers a new avenue into some interesting bicyclic structures.
Note: This post has been modified to correct the errors in the product structures and their associated InChIs and InChIKeys.
References
(1) Li, X.; Danishefsky, S. J., "Cyclobutenone as a Highly Reactive Dienophile: Expanding Upon Diels-Alder Paradigms," J. Am. Chem. Soc., 2010, 132, 11004-11005, DOI: 10.1021/ja1056888
InChIs
1: InChI=1/C4H4O/c5-4-2-1-3-4/h1-2H,3H2
InChIKey=DFLRGCFWSRELEL-UHFFFAOYAP
1prod: InChI=1/C8H10O/c9-8-5-6-3-1-2-4-7(6)8/h1-2,6-7H,3-5H2/t6-,7-/m0/s1
InChIKey=AYXQRXAAJYZWJJ-BQBZGAKWBC
2: InChI=1/C5H6O/c6-5-3-1-2-4-5/h1,3H,2,4H2
InChIKey=BZKFMUIJRXWWQK-UHFFFAOYAH
2prod: InChI=1/C9H12O/c10-9-6-5-7-3-1-2-4-8(7)9/h1-2,7-8H,3-6H2/t7-,8-/m0/s1
InChIKey=LOJATDUUSCWAOA-YUMQZZPRBU
3: InChI=1/C6H8O/c7-6-4-2-1-3-5-6/h2,4H,1,3,5H2
InChIKey=FWFSEYBSWVRWGL-UHFFFAOYAT
3prod: InChI=1/C10H14O/c11-10-7-3-5-8-4-1-2-6-9(8)10/h1-2,8-9H,3-7H2/t8-,9-/m0/s1
InChIKey=LFDGSLNQYSSFGI-IUCAKERBBQ