A 2013 study of oxalic acid 1 failed to uncover any tunneling between its conformations,1 despite observation of tunneling in other carboxylic acids (see this post). This was rationalized by computations which suggested rather high rearrangement barriers. Schreiner, Csaszar, and Allen have now re-examined oxalic acid using both experiments and computations and find what they call domino tunneling.2
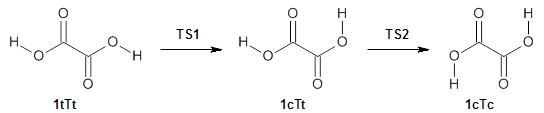
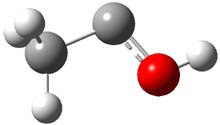
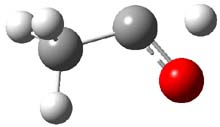
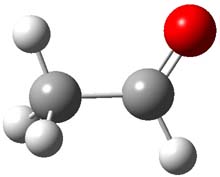
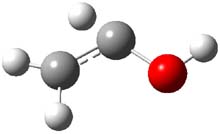
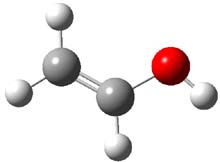
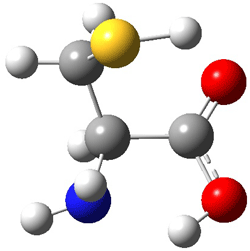
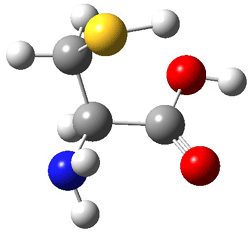
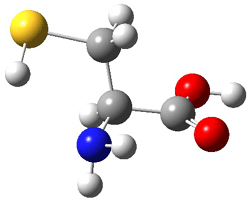
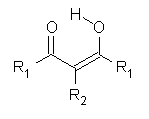
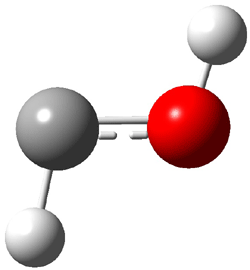
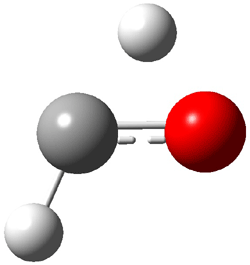
First, they determined the structures of the three conformations of 1 along with the two transition states interconnecting them using the focal point method. These geometries and relative energies are shown in Figure 1. The barrier for the two rearrangement steps are smaller than previous computations suggest, which suggests that tunneling may be possible.
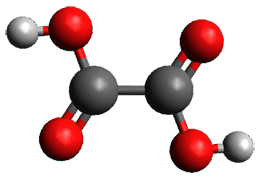
1tTt |
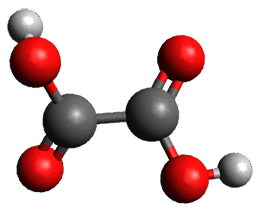
TS1 |
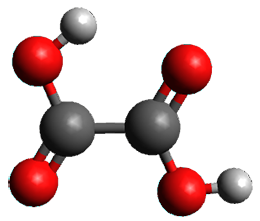
1cTt |
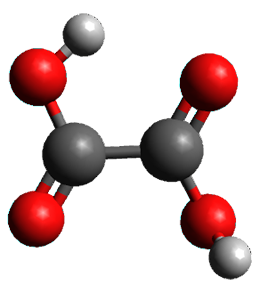
TS2 |
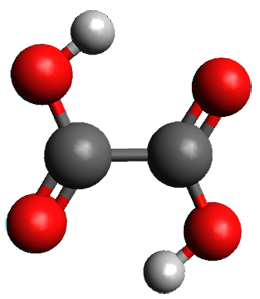
1cTc |
|
Figure 1. Geometries of the conformers of 1 and the TS for rearrangement and relative energies (kcal mol-1)
Placing oxalic acid in a neon matrix at 3 K and then exposing it to IR radiation populates the excited 1tTt conformation. This state then decays to both 1cTt and 1cTc, which can only happen through a tunneling process at this very cold temperature. Kinetic analysis indicates that there are two different rates for decay from both 1tTt and 1cTc, with the two rates associated with different types of sites within the matrix.
The intrinsic reaction paths for the two rearrangements: 1tTt → 1cTt and → 1cTc were obtained at MP2/aug-cc-pVTZ. Numerical integration and the WKB method yield similar half-lives: about 42 h for the first reaction and 23 h for the second reaction. These match up very well with the experimental half-lives from the fast matrix sites of 43 ± 4 h and 30 ± 20 h, respectively. Thus, the two steps take place sequentially via tunneling, like dominos falling over.
References
(1) Olbert-Majkut, A.; Ahokas, J.; Pettersson, M.; Lundell, J. "Visible Light-Driven Chemistry of Oxalic Acid in Solid Argon, Probed by Raman Spectroscopy," J. Phys. Chem. A 2013, 117, 1492-1502, DOI: 10.1021/jp311749z.
(2) Schreiner, P. R.; Wagner, J. P.; Reisenauer, H. P.; Gerbig, D.; Ley, D.; Sarka, J.; Császár, A. G.; Vaughn, A.; Allen, W. D. "Domino Tunneling," J. Am. Chem. Soc. 2015, 137, 7828-7834, DOI: 10.1021/jacs.5b03322.
InChIs
1: InChI=1S/C2H2O4/c3-1(4)2(5)6/h(H,3,4)(H,5,6)
InChIKey=MUBZPKHOEPUJKR-UHFFFAOYSA-N