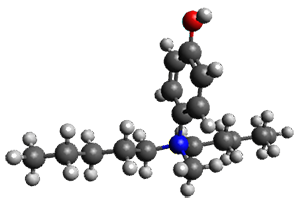
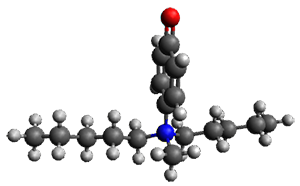
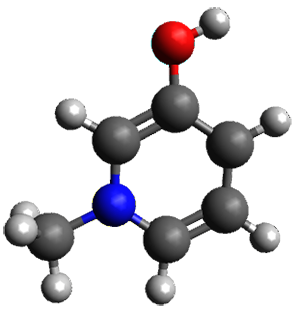
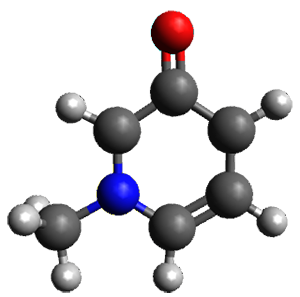
Houk and Vanderwal have examined the dyotropic rearrangement of an interesting class of polycyclic compounds using experimental and computational techniques.1 The parent reaction takes the bicyclo[2.2.2]octadiene 1 into the bicyclo[3.2.1]octadiene 3. The M06-2X/6-311+G(d,p)/B3LYP/6-31G(d) (with CPCM simulating xylene) geometries and relative energies are shown in Figure 1. The calculations indicate a stepwise mechanism, with an intervening zwitterion intermediate. The second step is rate determining.
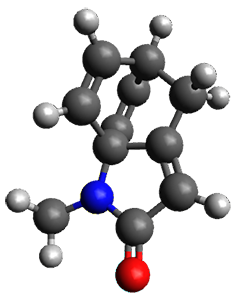
1 |
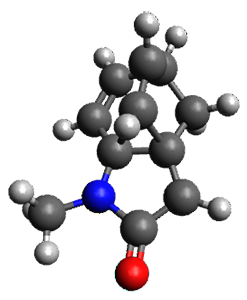
TS1 |
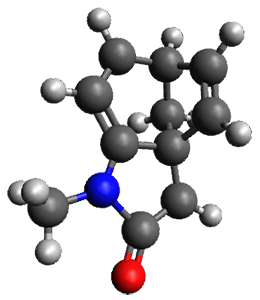
2 |
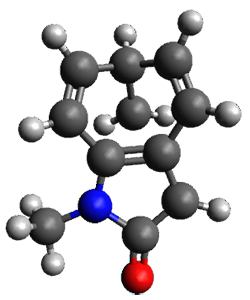
TS2 |
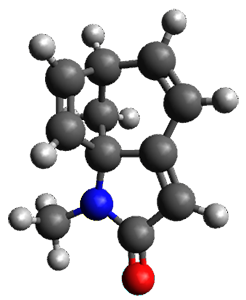 3 (-4.2) |
|
Figure 1. B3LYP/6-31G(d) and relative energies (kcal mol-1) at M06-2X/6-311+G(d,p).
Next they computed the activation barrier for the second TS for a series of substituted analogs of 1, with various electron withdrawing group as R1 and electron donating groups as R2, and compared them with the experimental rates.
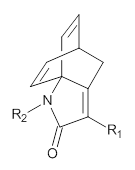
Further analysis was done by relating the charge distribution in these TSs with the relative rates, and they find a nice linear relationship between the charge and ln(krel). This led to the prediction that a cyano substituent would significantly activate the reaction, which was then confirmed by experiment. Another prediction of a rate enhancement with Lewis acids was also confirmed by experiment.
A last set of computations addressed the question of whether a ketone or lactone would also undergo this dyotropic rearrangement. The lactam turns out to have the lowest activation barrier by far.
References
(1) Pham, H. V.; Karns, A. S.; Vanderwal, C. D.; Houk, K. N. "Computational and Experimental Investigations of the Formal Dyotropic Rearrangements of Himbert Arene/Allene Cycloadducts," J. Am. Chem. Soc. 2015, 137, 6956-6964, DOI: 10.1021/jacs.5b03718.
InChIs
1: InChI=1S/C11H11NO/c1-12-10(13)7-9-6-8-2-4-11(9,12)5-3-8/h2-5,7-8H,6H2,1H3
InChIKey=MNYYUIQDOAXLTK-UHFFFAOYSA-N
3: InChI=1S/C11H11NO/c1-12-10(13)6-9-3-2-8-4-5-11(9,12)7-8/h2-6,8H,7H2,1H3
InChIKey=OHEBSZKLNGLATD-UHFFFAOYSA-N