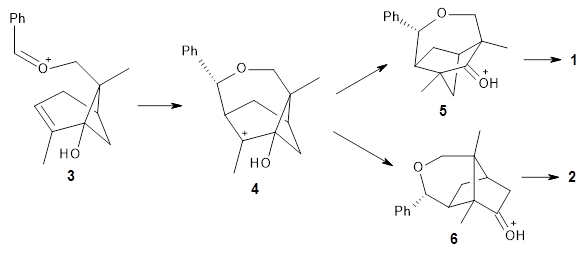
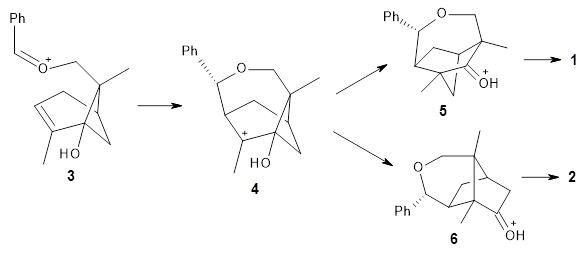
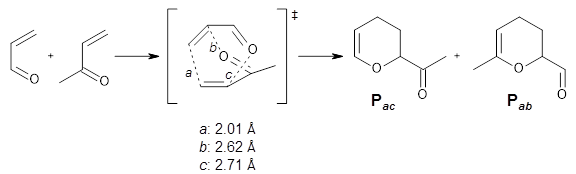
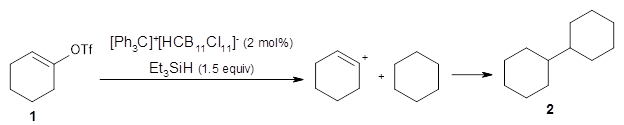A major topic of this blog has been the growing body of studies that demonstrate that dynamic effects can control reaction products (see these posts). Often these examples crop up with valley ridge inflection points. Another cause can be bispericyclic transition states, first discovered by Caramella et al for the dimerization of cyclopentadiene.1 The Houk group now reports on the first trispericyclic transition state.2
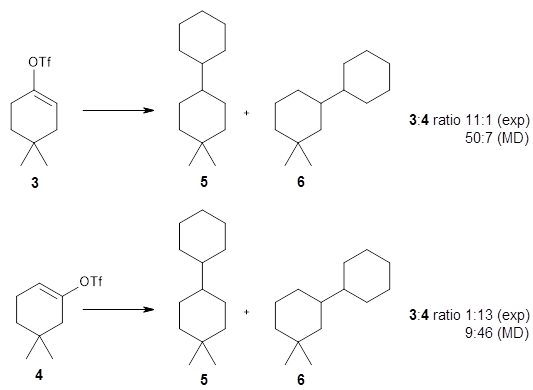
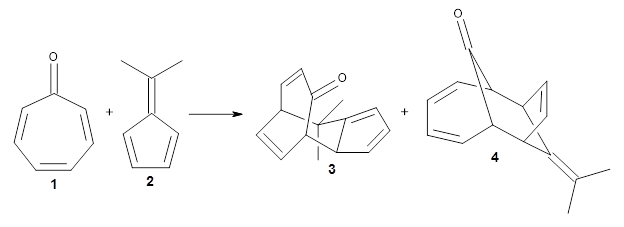
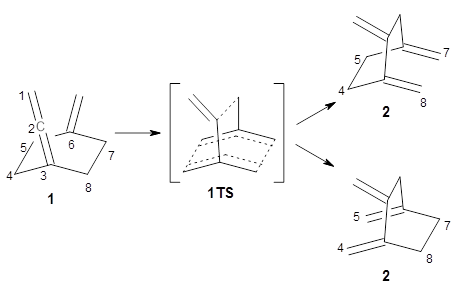
Using ωB97X-D/6-31G(d), they examined the reaction of the tropone derivative 1 with dimethylfulvene 2. Three possible products can arrive from different pericyclic reactions: 3, the [4+6] product; 4, the [6+4] product; and 5, the [8+2] product. The thermodynamic product is predicted to be 5, but it is only 1.2 kcal mol-1 lower in energy than 4 and 6.2 kcal mol-1 lower than 3.
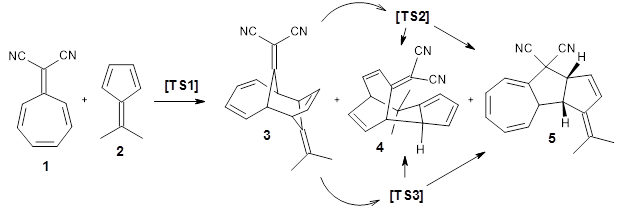
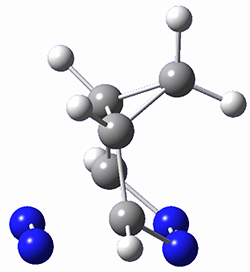
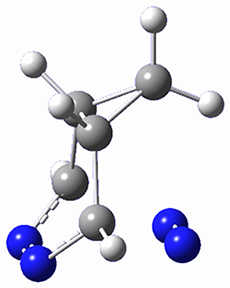
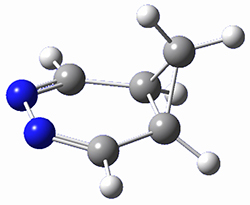
They identified one transition state originating from the reactants TS1. Hypothesizing that it would be trispericyclic, they performed a molecular dynamics study with trajectories starting from TS1. They ran a total of 142 trajectories, and 87% led to 3, 3% led to 4, and 3% led to 5. This demonstrates the unusual nature of TS1 and the dynamic effects on this reaction surface.
|
|
|
|
|
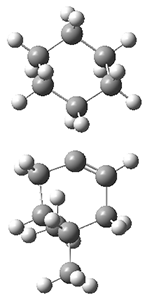
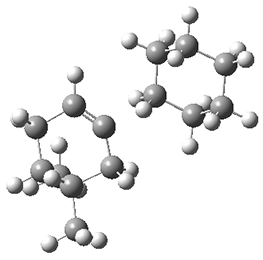
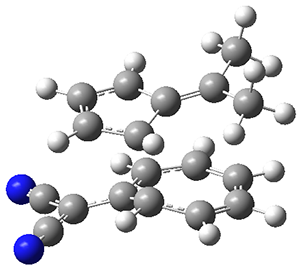 TS3 |
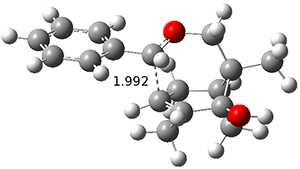
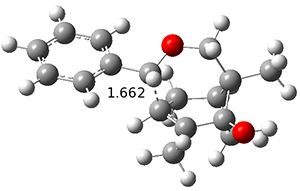
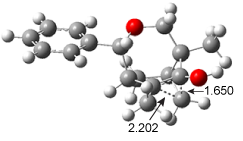
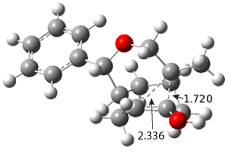
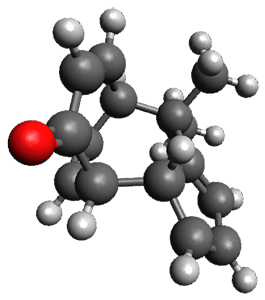
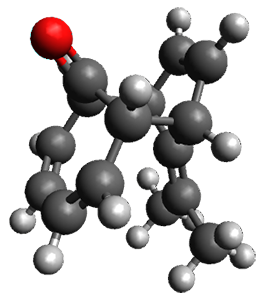
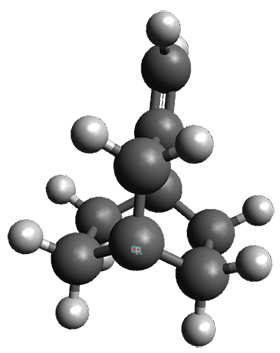
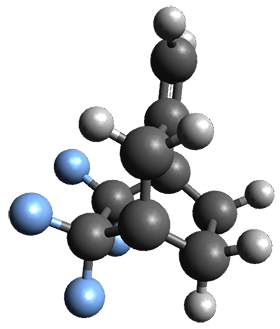
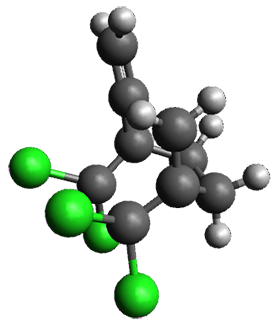
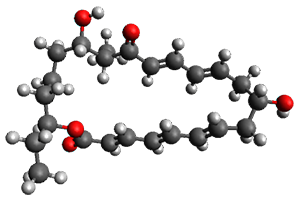
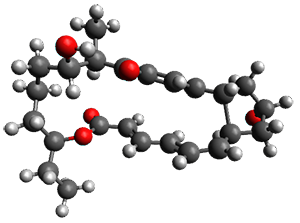
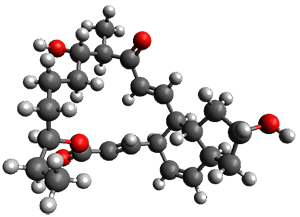
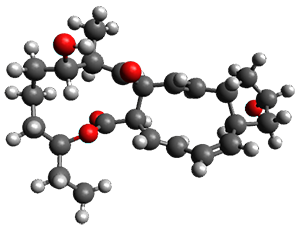
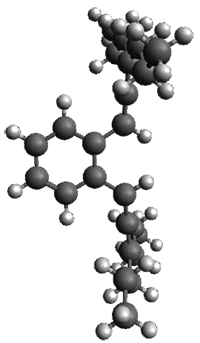
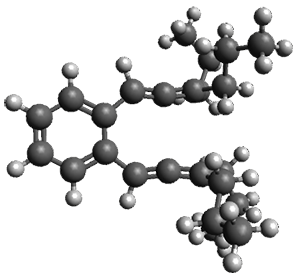
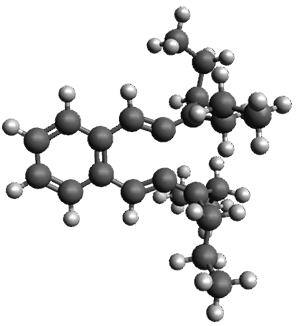
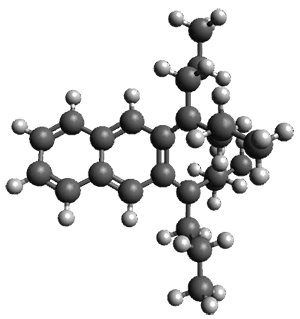
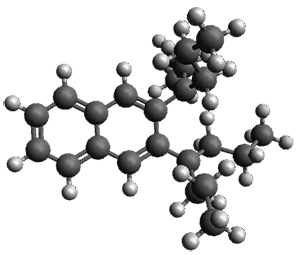
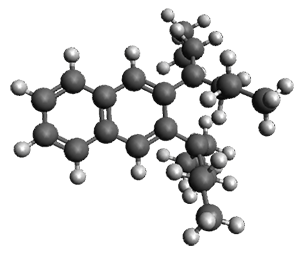
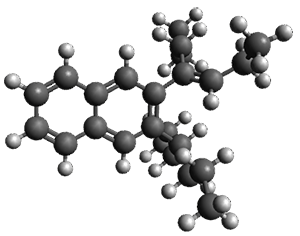
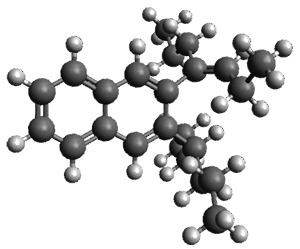
Figure 1. ωB97X-D/6-31G(d) optimized geometries of TS1-TS3.
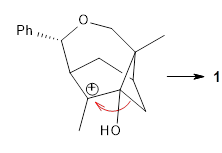
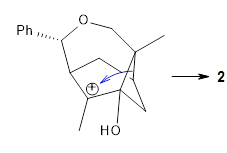
Additionally, there are two different Cope rearrangements (through TS2 and TS3) that convert 3 into 4 and 5. Some trajectories can pass from TS1 and then directly through either TS2 or TS3 and these give rise to products 4 and 5. In other words, some trajectories will pass from a trispericyclic transition state and then through a bispericyclic transition state before ending in product.
References
1. Caramella, P.; Quadrelli, P.; Toma, L., “An Unexpected Bispericyclic Transition Structure Leading to 4+2 and 2+4 Cycloadducts in the Endo Dimerization of Cyclopentadiene.” J. Am. Chem. Soc. 2002, 124, 1130-1131, DOI: 10.1021/ja016622h
2. Xue, X.-S.; Jamieson, C. S.; Garcia-Borràs, M.; Dong, X.; Yang, Z.; Houk, K. N., “Ambimodal Trispericyclic Transition State and Dynamic Control of Periselectivity.” J. Am. Chem. Soc. 2019, 141, 1217-1221, DOI: 10.1021/jacs.8b12674.
InChIs
1: InChI=1S/C10H6N2/c11-7-10(8-12)9-5-3-1-2-4-6-9/h1-6H
InChIKey=KAWLLELUFONBGI-UHFFFAOYSA-N
2: InChI=1S/C8H10/c1-7(2)8-5-3-4-6-8/h3-6H,1-2H3
InChIKey=WXACXMWYHXOSIX-UHFFFAOYSA-N
3: InChI=1S/C18H16N2/c1-11(2)17-15-7-8-16(17)14-6-4-3-5-13(15)18(14)12(9-19)10-20/h3-8,13-16H,1-2H3
InChIKey=DRPXVBLNTKGMTB-UHFFFAOYSA-N
4: InChI=1S/C18H16N2/c1-18(2)13-6-8-14(12(10-19)11-20)15(9-7-13)16-4-3-5-17(16)18/h3-9,13,15-16H,1-2H3
InChIKey=FSIPGNLAWKVXDD-UHFFFAOYSA-N
5: InChI=1S/C18H16N2/c1-12(2)13-8-9-16-17(13)14-6-4-3-5-7-15(14)18(16,10-19)11-20/h3-9,14,16-17H,1-2H3/t14?,16-,17-/m1/s1
InChIKey=SYLWEGLODFLARZ-VNCLPFQGSA-N