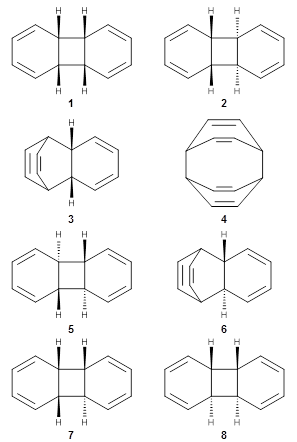
Pericyclic reactions remain a fruitful area of research despite the seminal publication of the Woodward-Hoffmann rules decades ago. Here are two related papers of pericyclic reactions that violate the Woodward-Hoffmann rules.
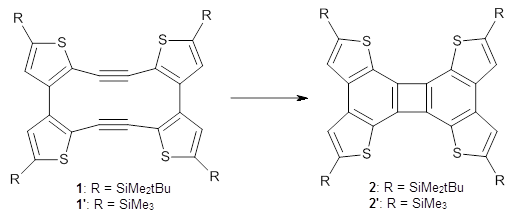
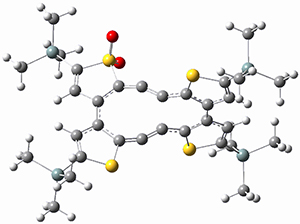
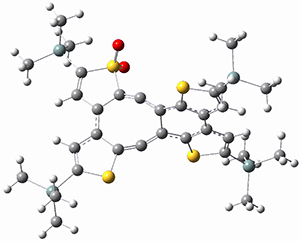
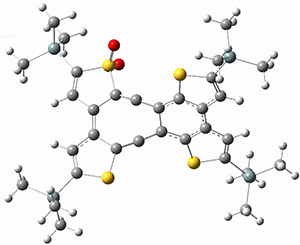
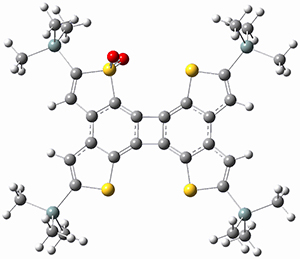
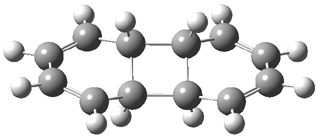
First, Solomek, Ravat, Mou, Kertesz, and Jurícek reported on the thermal and photochemical electrocyclization reaction of diphenylcetherene 1a.1 Though they were not able to directly detect the intermediate 2, through careful examination of the photochemical reaction, they were able to infer that the thermal cyclization goes via the formally forbidden conrotatory pathway (see Scheme 1).
Scheme 2.
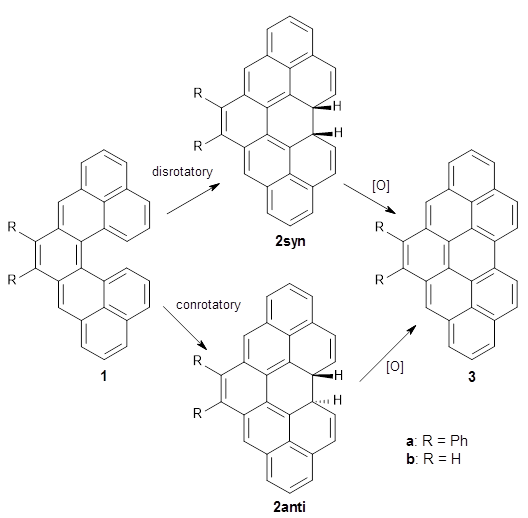
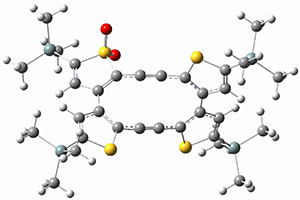
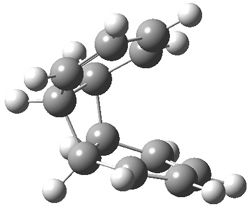
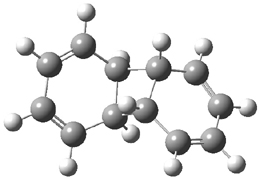
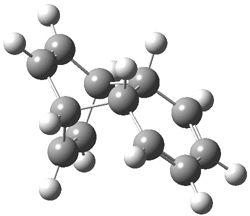
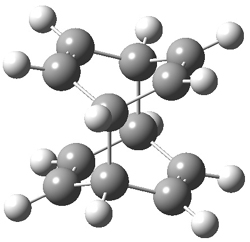
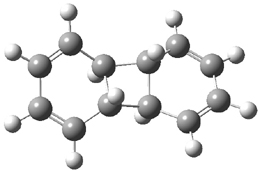
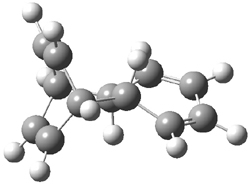
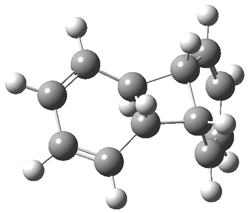
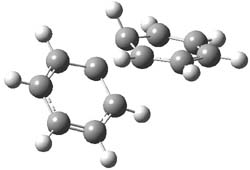
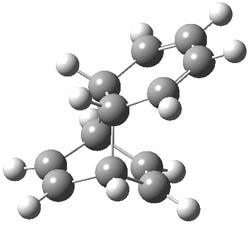
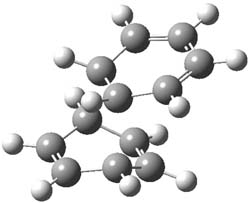
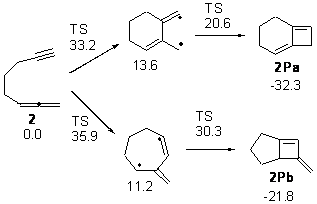
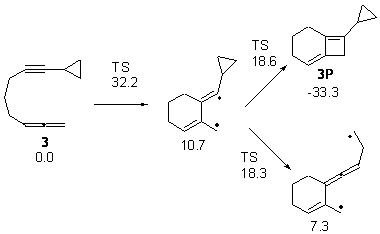
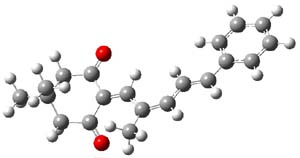
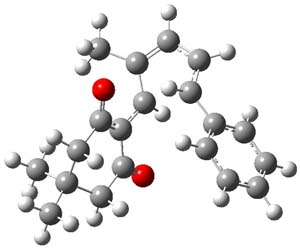
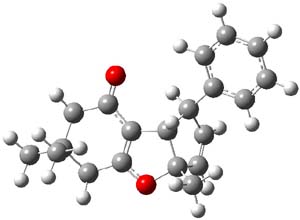
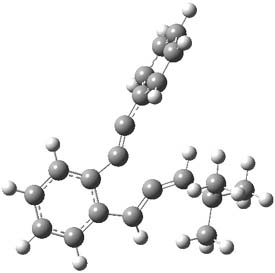
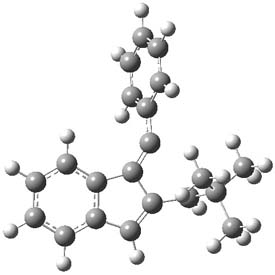
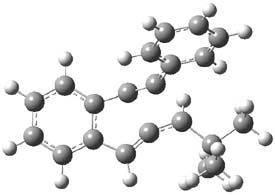
Kinetic studies estimate the activation barrier is 14.1 kcal mol-1. They performed DFT computations of the parent 1b using a variety of functionals with both restricted and unrestricted wavefunctions. The allowed pathway to 2syn is predicted to be greater than 27 kcal mol-1, while the formally forbidden pathway to 2anti is estimated to have a lower barrier of about 23 kcal mol-1. The two transition states for these different pathways are shown in Figure 1, and the sterics that force a helical structure to 1 help make the forbidden pathway more favorable.
|
|
|
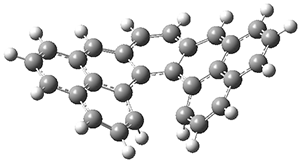
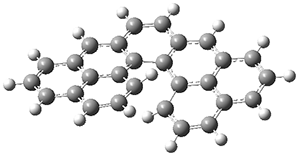
Figure 1. (U)B3LYP/6-31G optimized geometries of the transition states taking 1 into 2.
Nonetheless, all of the DFT computations significantly overestimate the activation barrier. The authors make the case that a low-lying singlet excited state results in an early conical intersection that reduces the symmetry from C2 to C1. In this lower symmetry pathway, all of the states can mix, leading to a lower barrier. However, since DFT is intrinsically a single Slater configuration, the mixing of the other states is not accounted for, leading to the overestimated barrier height.
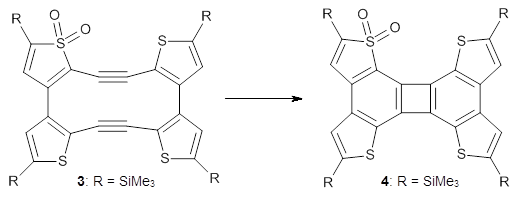
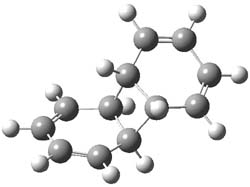
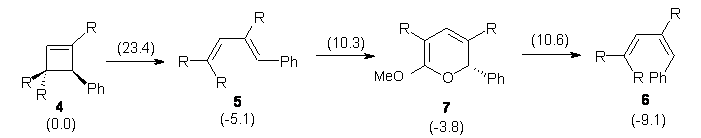
In a follow up study, this group examined the thermal and photo cyclization of 13,14-dimethylcethrene 4.
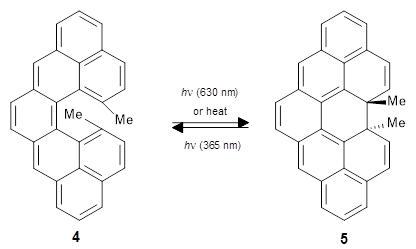
Also of interest is the singlet-triplet gap of 1 and 4. The DFT computed ΔEST is about 10 kcal mol-1 for 4, larger than the computed value of 6 kcal mol-1 for 1b. The EPR of 1b does show a signal while that of 4 has no signal. To assess the role of the methyl group, they computed the singlet triplet gaps for 1b and 4 at two different geometries: where the distance between the carbons bearing the methyl groups is that in 1b (3.03 Å) and in 4 (3.37 Å). The lengthening of this distance by the methyl substituents is due to increased helical twist in 4 than in 1b. For 1b, the gap increases with twisting, from 7.1 to 8.3 kcal mol-1, while for 4 the gap increases by 1.8 kcal mol-1 with the increased twisting. This change is less than the effect of methyl substitution, which increases the gap by 2.2 kcal mol-1 at the shorter distance and 2.8 kcal mol-1 at the longer distance. Thus, the electronic (orbital) effect of methyl substitution affects the singlet-triplet gap more than the geometric twisting.
References
1) Šolomek, T.; Ravat, P.; Mou, Z.; Kertesz, M.; Juríček, M., "Cethrene: The Chameleon of Woodward–Hoffmann Rules." J. Org. Chem. 2018, 83, 4769-4774, DOI: 10.1021/acs.joc.8b00656.
2) Ravat, P.; Šolomek, T.; Häussinger, D.; Blacque, O.; Juríček, M., "Dimethylcethrene: A Chiroptical Diradicaloid Photoswitch." J. Am. Chem. Soc. 2018, 140, 10839-10847, DOI: 10.1021/jacs.8b05465.
InChIs
1b: InChI=1S/C28H16/c1-5-17-7-3-11-23-25(17)19(9-1)15-21-13-14-22-16-20-10-2-6-18-8-4-12-24(26(18)20)28(22)27(21)23/h1-16H
InChIKey=GBMHAGKZRAVBDO-UHFFFAOYSA-N
4: InChI=1S/C30H20/c1-17-9-11-19-5-3-7-21-15-23-13-14-24-16-22-8-4-6-20-12-10-18(2)26(28(20)22)30(24)29(23)25(17)27(19)21/h3-16H,1-2H3
InChIKey=MXTVFWTUCPRNIW-UHFFFAOYSA-N
5: nChI=1S/C30H20/c1-29-13-11-17-5-3-7-19-15-21-9-10-22-16-20-8-4-6-18-12-14-30(29,2)28(24(18)20)26(22)25(21)27(29)23(17)19/h3-16H,1-2H3/t29-,30-/m0/s1
InChIKey=SUMMGEBJORQMAI-KYJUHHDHSA-N