Castro and Karney1 previously predicted a Möbius aromatic transition state for the π-bond shift in [12]annulene (see Chapter 2.4.3.1), a process they termed “twist-couple bond shifting”. In late 2006 they turned their attention to the conformational surface of [16]annulene, searching again for Möbius aromatic ground or transition states.2
Oth synthesized [16]annulene by the photolysis of cycloctatetraene dimer. He observed two isomers 1a and 2a in a 83:17 ratio3 at -140 °C, with a barrier4 of 10.3 kcal mol-1 separating them. The 1H NMR spectrum at -30 °C shows only one signal. The equivalence of all of the protons implicates rapid conformational changes and bond shifting, as suggested in Scheme 1. Also noted was that these conversions, including the configuration change from 1 to 2, have barriers much lower than for the electrocyclization of Reaction 1 of about 22 kcal mol-1.5
|
Scheme 1

|

|
Reaction 1
|
Following on the results from their [12]annulene study, Castro and Karney optimized geometries at BH&HLYP/6-311+G(d,p). Since, as we discussed in Chapter 2.4.3.1, relative energies of annulene conformations are very sensitive to the computational method and basis set, they determined estimated CCSD(T)/cc-pVDZ energies, which I will call Eest, according to a prescription proposed by Bally and MacMahon,6 namely
|
Eest = E(HF/cc-pVDZ) +
|
| |
Ecorr(MP2/cc-pvDZ)
|
Ecorr(CCSD(T)/6-31G(d))
|
|
Ecorr(MP2/6-31G(d))
|
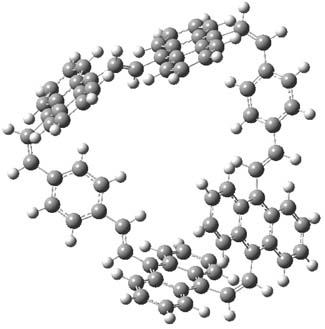
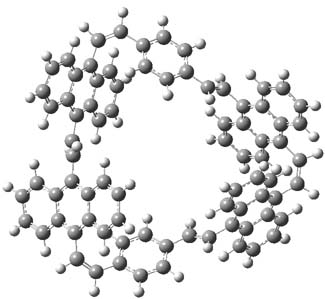
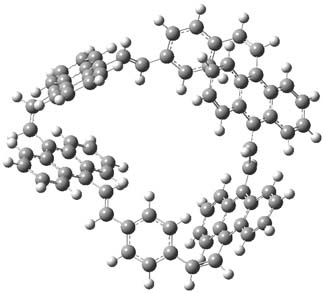
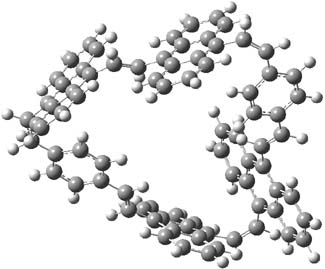
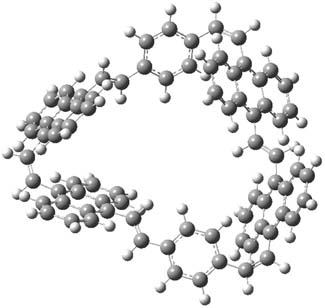
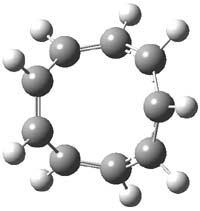
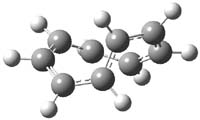
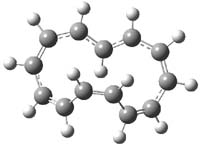
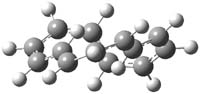
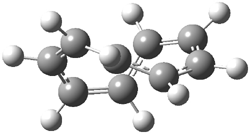
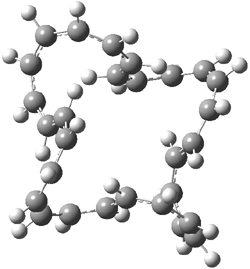
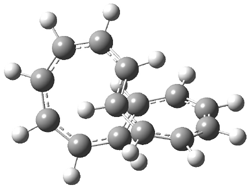
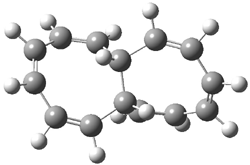
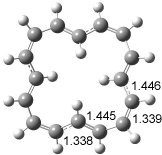
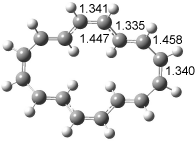
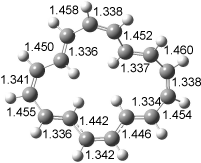
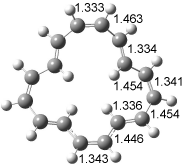
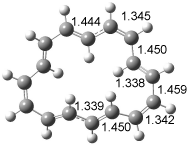
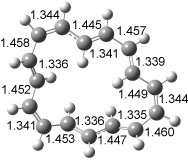
The optimized structures of 1a and 2a are drawn in Figure 1. Both molecules are not planar, their bond lengths are clearly alternating, and their NICS(0) values are +6.4 ppm (1a) and +7.3 ppm (2a), all evidence that neither molecule is aromatic. 1a is predicted to be 0.8 kcal mol-1 lower in energy than 2a, consistent with experiment.
Figure 1. BH&HLYP/6-311+G(d,p) optimized geometries and relative energies (kcal mol-1) based on Eest.2
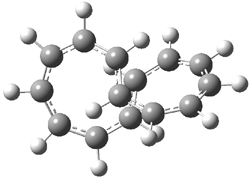
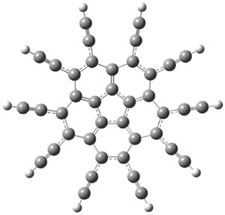
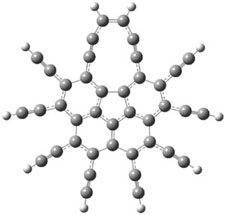
The conformational change 1a → 1a’ is a multi-step process. This is in contrast to [12]annulene where this change occurs via a concerted mechanism. So, 1a first converts to 1c through a barrier of 7.9 kcal mol-1. The path now splits; 1b can next be formed with a barrier of 9.4 kcal mol-1 to give 1c’ or 1d can be formed through a barrier of 7.7 kcal mol-1 to produce 1c’. 1c’ converts to 1a’ with a barrier of 7.9 kcal mol-1. The structures of the intermediates and their relative energies are shown in Figure 1.
The conversion of 1 to 2 takes place through the transition state TS-1c2b that actually connects isomer 1c to 2b. This structure, shown in Figure 1, exhibits little bond alternation and has a NICS(0) value of -14.2, both strongly suggestive of Möbius aromatic character. Aromaticity should also imply energetic stabilization; TS-1c2b lies only 13.7 kcal mol-1 above 1a. This barrier is less than that predicted for the twist-coupled bond shift in either [10]annulene or [12]annulene.
The highest barrier for the various interconversions indicated in Scheme 1 is the barrier associated with TS-1c2b. This barrier (13.7 kcal mol-1) is significantly lower that the activation energy for Reaction 1 (22 kcal mol-1). These computations confirm that the scrambling of the protons of [16]annulene is due to the rapid rearrangements of Scheme 1. Furthermore, the computations demonstrate that the key step is a twist-coupled bond shift that is facilitated by the Möbius aromatic character of its transition state.
Since the configuration change in [12]- and [16]annulene proceeds with a bond-shifting Möbius aromatic bond shifting transition state, might not the configuration change of [14]annulene proceed through a Möbius antiaromatic bond shifting transition state? In 2007, Castro and Karney7 answered this question in the affirmative.
Consistent with their previous studies, geometries were optimized at UBH&HLYP/6-311+G**. The unrestricted method is necessary since the expected antiaromatic transition state will have singlet radical character. In order to obtain reasonable energies, CASPT2(14,14)/cc-pVDZ single-point computations were employed.
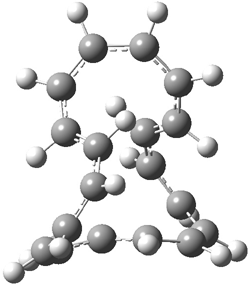
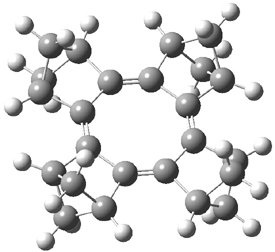
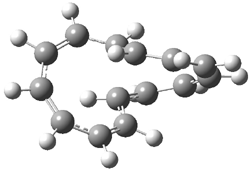
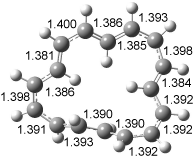
[14]annulene must undergo two conformational changes (3a-c) before the bond shift/configuration change can occur through transition state 4 to give 5. Note that this process changes the number of cis and trans double bonds. This overall process is shown in Figure 2. The optimized structures of 3c, 4, and 5 are shown in Figure 3.
Figure 2. CASPT2(14,14)/cc-pVDZ// UBH&HLYP/6-311+G** relative energies of stable structures along the pathway for configuration change of [14]annulene.
The computed barrier for the configuration change (through 4) is computed to be 19.3 kcal mol-1, in very reasonable agreement with the experimental value4 of 21.3 kcal mol-1.
Figure 3. UBH&HLYP/6-311+G** optimized geometries of 3c, 4, and 5.7
Based on its magnetic properties, transition state 4 has decided antiaromatic character. Its computed NICS(0) value is +19.0 ppm. Compare this to the NICS(0) values for 3a and 5 of -8.0 and -5.0 ppm, respectively. In addition, the computed chemical shifts of the two interior protons are very downfield, 26.4 and 26.7 ppm.
InChI
1: InChI=1/C16H16/c1-2-4-6-8-10-12-14-16-15-13-11-9-7-5-3-1/h1-16H/b2-1-,3-1-,4-2+,5-3+,6-4+,7-5+,8-6-,9-7-,10-8-,11-9-,12-10+,13-11+,14-12+,15-13+,16-14-,16-15-
2: InChI=1/C16H16/c1-2-4-6-8-10-12-14-16-15-13-11-9-7-5-3-1/h1-16H/b2-1-,3-1-,4-2+,5-3+,6-4+,7-5+,8-6-,9-7+,10-8+,11-9+,12-10+,13-11-,14-12-,15-13-,16-14+,16-15+
3: InChI=1/C14H14/c1-2-4-6-8-10-12-14-13-11-9-7-5-3-1/h1-14H/b2-1-,3-1-,4-2+,5-3+,6-4+,7-5+,8-6-,9-7-,10-8+,11-9+,12-10+,13-11+,14-12-,14-13-
5: InChI=1/C14H14/c1-2-4-6-8-10-12-14-13-11-9-7-5-3-1/h1-14H/b2-1-,3-1-,4-2-,5-3+,6-4+,7-5+,8-6+,9-7-,10-8-,11-9-,12-10-,13-11-,14-12+,14-13+
References
(1) Castro, C.; Karney, W. L.; Valencia, M. A.; Vu, C. M. H.; Pemberton, R. P., “Möbius Aromaticity in [12]Annulene: Cis-Trans Isomerization via Twist-Coupled Bond Shifting,” J. Am. Chem. Soc. 2005, 127, 9704-9705, DOI: 10.1021/ja052447j.
(2) Pemberton, R. P.; McShane, C. M.; Castro, C.; Karney, W. L., “Dynamic Processes in [16]Annulene: Change,” J. Am. Chem. Soc. 2006, 128, 16692-16700, DOI: 10.1021/ja066152x
(3) Oth, J. F. M.; Anthoine, G.; Gilles, J.-M., “Le dianion du [16] annulene,” Tetrahedron Lett. 1968, 9, 6265-6270, DOI: 10.1016/S0040-4039(00)75449-9.
(4) Oth, J. F. M., “Conformational Mobility and Fast Bond Shift in the Annulenes,” Pure Appl. Chem. 1971, 25, 573-622.
(5) Schroeder, G.; Martin, W.; Oth, J. F. M., “Thermal and Photochemical Behavior of a [16]Annulene,” Angew. Chem. Int. Ed. Engl. 1967, 6, 870-871, DOI: 10.1002/anie.196708701.
(6) Matzinger, S.; Bally, T.; Patterson, E. V.; McMahon, R. J., “The C7H6 Potential Energy Surface Revisited: Relative Energies and IR Assignment,” J. Am. Chem. Soc. 1996, 118, 1535-1542, DOI: 10.1021/ja953579n.
(7) Moll, J. F.; Pemberton, R. P.; Gutierrez, M. G.; Castro, C.; Karney, W. L., “Configuration Change in [14]Annulene Requires Möbius Antiaromatic Bond Shifting,” J. Am. Chem. Soc. 2007, 129, 274-275, DOI: 10.1021/ja0678469.