Here are a couple of articles describing computational approaches to catalytic enantioselective reactions using variations upon the classic proline-catalyzed aldol reaction of List and Barbas1 that started the whole parade. I have discussed the major computational papers on that system in my book (Chapter 5.3).
Yang and Wong2 investigated the proline-catalyzed nitro-Michael reaction, looking at four examples, two with aldehydes and two with ketones (Reactions 1-4).
|
Reaction 1
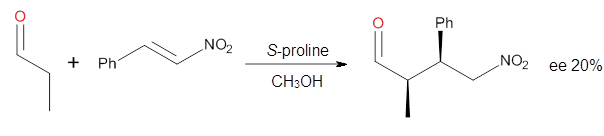
|
|
Reaction 2
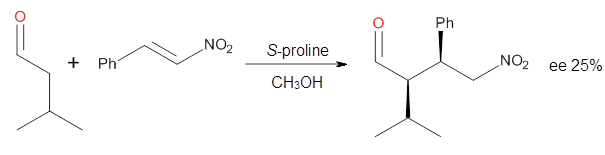
|
|
Reaction 3
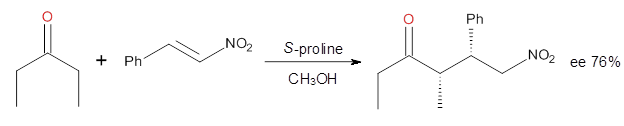
|
|
Reaction 4
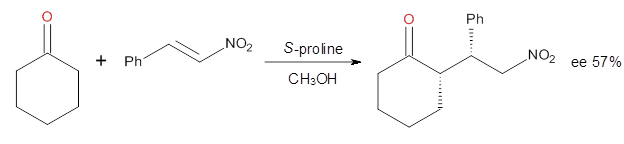
|
These four reactions were examined atMP2/311+G**//M06-2x/6-31G**, and PCM was also applied. The key element of this study is that they examined two different types of transition states: (a) based on the Houk-List model involving a hydrogen bond and (b) an electrostatic based model with no hydrogen bond. These are sketched in Scheme 1. For each of the reactions 1-4 there are 8 located transition states differing in the orientation of the attack on to the syn or anti enamine.
Scheme 1. TS models
|
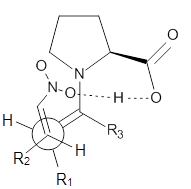
Model A
|
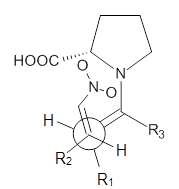
Model B
|
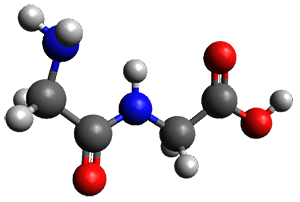
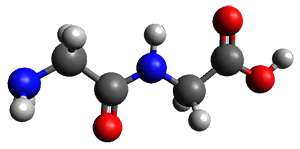
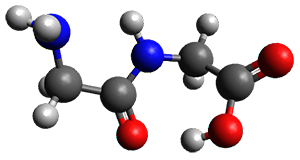
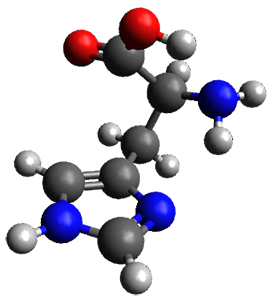
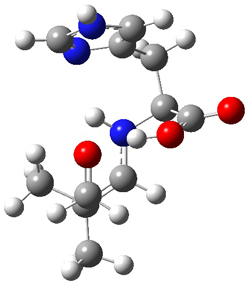
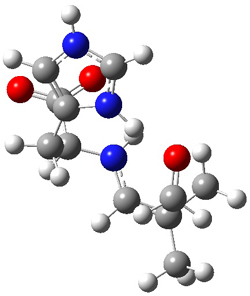
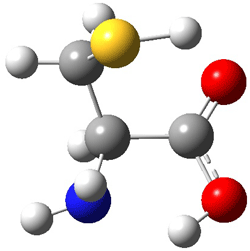
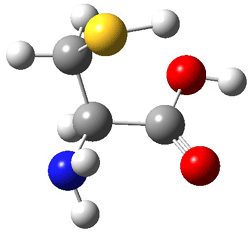
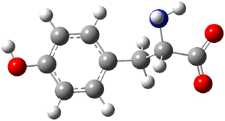
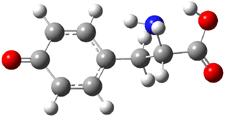
The two lowest energy TS are shown in Figure 1. TS1-β1-RS is the lowest TS and it leads to the major enantiomer. The second lowest TS, TS1-β3-SR, lies 2.9 kJ mol-1 above the other TS, and it leads to the minor enantiomer. This lowest TS is of the Houk-List type (Model A) while the other TS is of the Model B type. The enthalpies of activation suggest an ee of 54%, in reasonable
agreement with experiment.
Figure 1. M06-2x/6-31G** optimized geometries of TS1-β1-RS and TS1-β1-RS.
The computations of the other three reactions are equally good in terms of agreement with experiment, and importantly the computations indicate the reversal of stereoselection between the aldehydes and the ketones. These computations clearly implicate both the Houk-List and the non-hydrogen bonding TSs in the catalyzed Michael additions.
Houk in collaboration with Scheffler and Mahrwald investigate the use of histidine as a catalyst for the asymmetricaldol reaction.3 Examples of the histidine-catalyzed aldol are shown in Reactions 5-7.
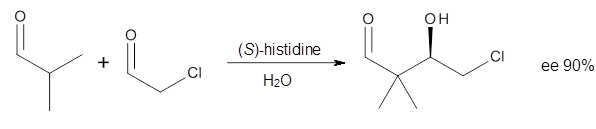
|
Reaction 5
|
|
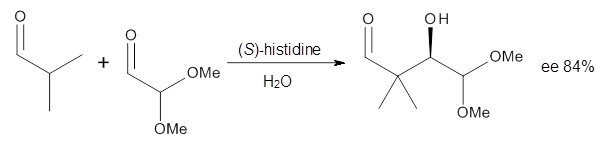
|
Reaction 6
|
|
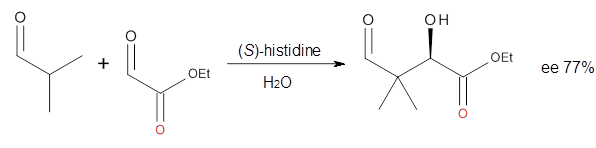
|
Reaction 7
|
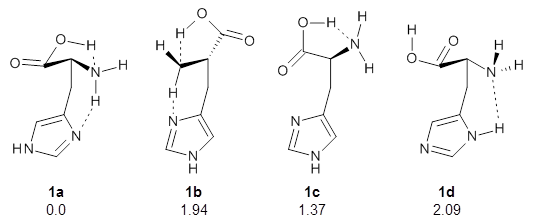
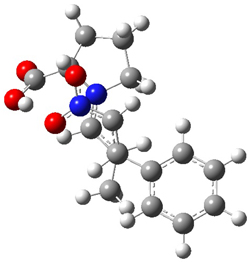
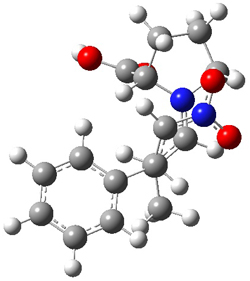
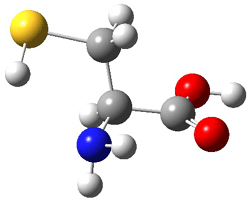
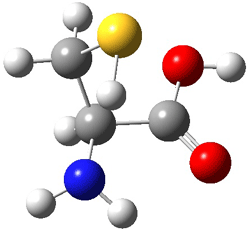
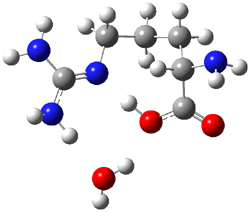
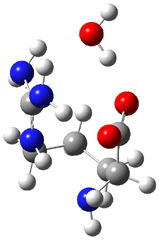
The interesting twist here is whether the imidazole can also be involved in hydrogen bonding to the acceptor carbonyl group, serving the purpose of the carboxylic acid group in the Houk-List TS when proline is the catalyst (Model A). The transition states for these and a few other reactions were computed at M06-2x/6-31+G(d,p) including with the SMD continuum solvent model for water. The two lowest energy TSs for the reaction of isobutyraldehde and formaldehyde are shown in Figure 2; TS1 has the carboxylic acid group as the hydrogen donor while the imidazole is the donor in TS2. Of note is that these two TS are isoenergetic, indicating that both modes of stabilization are at play with histidine as the catalyst.
Figure 2. M06-2x/6-31+G(d,p) geometries of the two lowest energy TSs for the reaction of isobutyraldehde and formaldehyde catalyzed by histidine.
The possible TSs for Reactions 5-7 were also located. For example, with Reaction 5, the lowest energy TS involves the imidazole as the hydrogen donor and it leads to the major product. The lowest energy TS that leads to the minor product involves the carboxylic acid as the donor. The computed ee’s for Reactions 5-7 are in very good, if not excellent, agreement with the experimental values. The study should spur further activity in which one might tune the stereoselectivity by using catalysts with multiple binding opportunities.
References
(1) List, B.; Lerner, R. A.; Barbas, C. F., III; "Proline-Catalyzed Direct Asymmetric Aldol Reactions," J. Am. Chem. Soc., 2000, 122, 2395-2396, DOI: 10.1021/ja994280y.
(2) Yang, H.; Wong, M. W. "(S)-Proline-catalyzed nitro-Michael reactions: towards a better understanding of the catalytic mechanism and enantioselectivity," Org. Biomol. Chem.,
2012, 10, 3229-3235, DOI: 10.1039/C2OB06993H
(3) Lam, Y.-h.; Houk, K. N.; Scheffler, U.; Mahrwald, R. "Stereoselectivities of Histidine-Catalyzed Asymmetric Aldol Additions and Contrasts with Proline Catalysis: A Quantum Mechanical Analysis," J. Am. Chem. Soc. 2012, 134, 6286-6295, DOI: 10.1021/ja2118392