The role of dispersion in organic chemistry has been slowly recognized as being quite critical in a variety of systems. I have blogged on this subject many times, discussing new methods for properly treating dispersion within quantum computations along with a variety of molecular systems where dispersion plays a critical role. Schreiner1 has recently published a very nice review of molecular systems where dispersion is a key component towards understanding structure and/or properties.
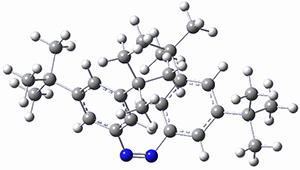
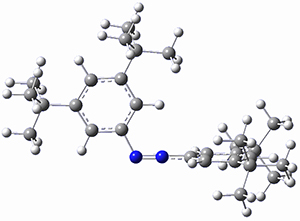
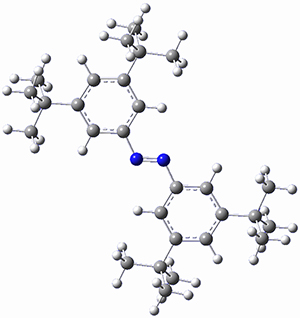
In a similar vein, Wegner and coworkers have examined the Z to E transition of azobenzene systems (1a-g → 2a-g) using both experiment and computation.2 They excited the azobenzenes to the Z conformation and then monitored the rate for conversion to the E conformation. In addition they optimized the geometries of the two conformers and the transition state for their interconversion at both B3LYP/6-311G(d,p) and B3LYP-D3/6-311G(d,p). The optimized structure of the t-butyl-substituted system is shown in Figure 1.
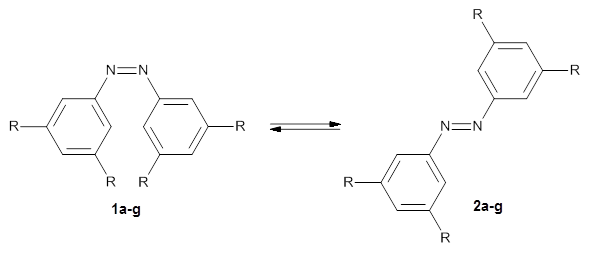
a: R=H; b: R=tBu; c: R=Me; d: R=iPr; e: R=Cyclohexyl; f: R=Adamantyl; g: R=Ph
Figure 1. B3LYP-D3/6-311G(d,p) optimized geometries of 1a, 2a, and the TS connecting them.
The experiment finds that the largest activation barriers are for the adamantly 1f and t-butyl 1b azobenzenes, while the lowest barriers are for the parent 1a and methylated 1c azobenzenes.
The trends in these barriers are not reproduced at B3LYP but are reproduced at B3LYP-D3. This suggests that dispersion is playing a role. In the Z conformations, the two phenyl groups are close together, and if appropriately substituted with bulky substituents, contrary to what might be traditionally thought, the steric bulk does not destabilize the Z form but actually serves to increase the dispersion stabilization between these groups. This leads to a higher barrier for conversion from the Z conformer to the E conformer with increasing steric bulk.
References
(1) Wagner, J. P.; Schreiner, P. R. "London Dispersion in Molecular Chemistry—Reconsidering Steric Effects," Angew. Chem. Int. Ed. 2015, 54, 12274-12296, DOI: 10.1002/anie.201503476.
(2) Schweighauser, L.; Strauss, M. A.; Bellotto, S.; Wegner, H. A. "Attraction or Repulsion? London Dispersion Forces Control Azobenzene Switches," Angew. Chem. Int. Ed. 2015, 54, 13436-13439, DOI: 10.1002/anie.201506126.
InChIs
1b: InChI=1S/C28H42N2/c1-25(2,3)19-13-20(26(4,5)6)16-23(15-19)29-30-24-17-21(27(7,8)9)14-22(18-24)28(10,11)12/h13-18H,1-12H3/b30-29-
InChIKey=SOCNVTNVHBWFKC-FLWNBWAVSA-N
2b: InChI=1S/C28H42N2/c1-25(2,3)19-13-20(26(4,5)6)16-23(15-19)29-30-24-17-21(27(7,8)9)14-22(18-24)28(10,11)12/h13-18H,1-12H3/b30-29+
InChIKey=SOCNVTNVHBWFKC-QVIHXGFCSA-N