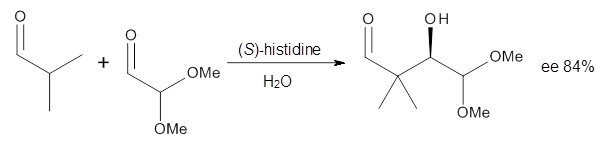
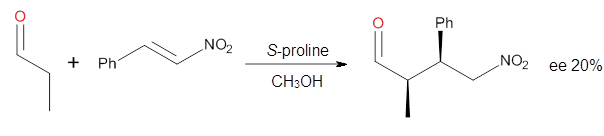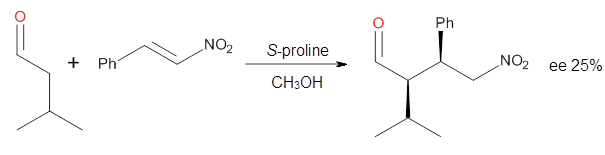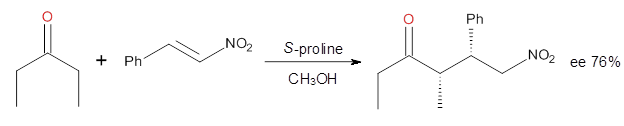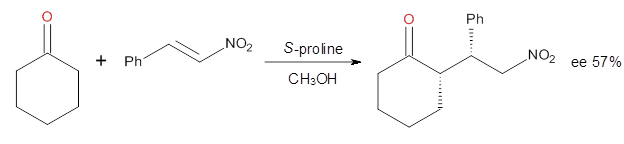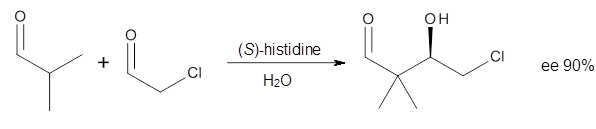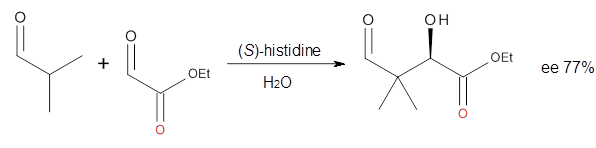Cinchona alkaloids cat catalyze reactions, such as shown in Reaction 1. Wynberg1 proposed a model to explain the reaction, shown in Scheme 1, based on NMR. Grayson and Houk have now used DFT computations to show that the mechanism actually reverses the arrangements of the substrates.2
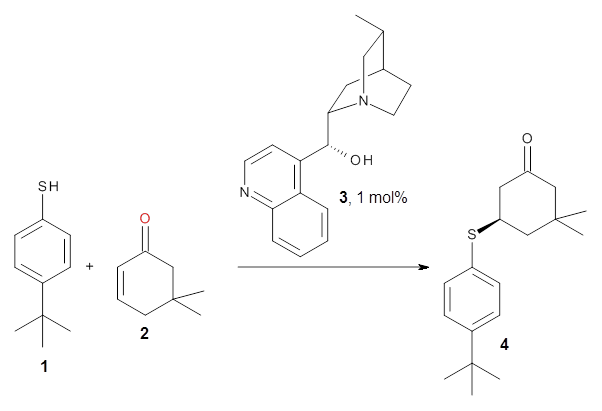
|
Reaction 1 |
Scheme 1.
|
|
|
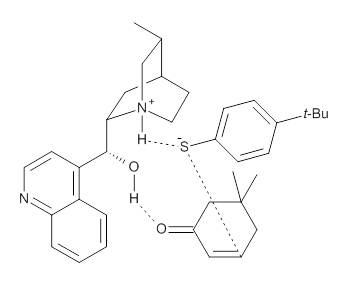
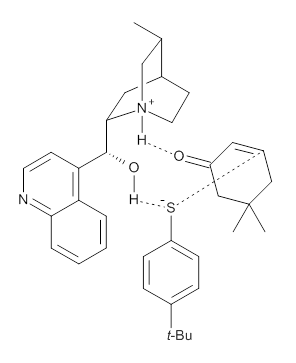
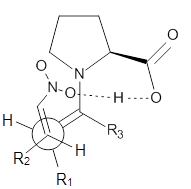
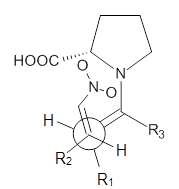
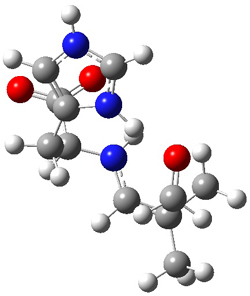
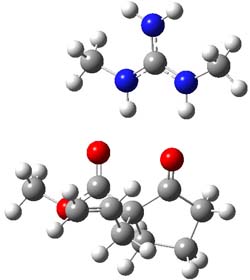
M06-2X/def2-TZVPP−IEFPCM(benzene)//M06-2X/6-31G(d)−IEFPCM(benzene) computations show that the precomplex of catalyst 3 with nucleophile 1 and Michael acceptor 2 is consistent with Wynberg’s model. The alternate precomplex is 5.6 kcal mol-1 higher in energy. These precomplexes are shown in Figure 1.
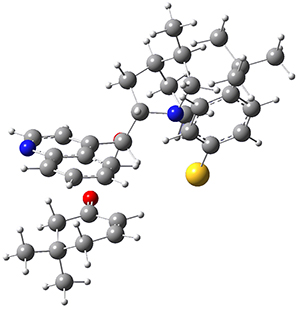
Wynberg precomplex |
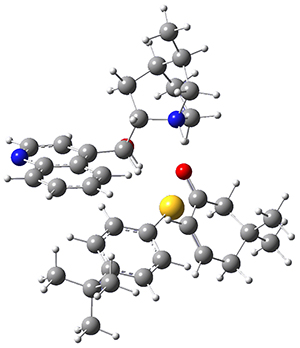
Grayson/Houk precomplex |
Figure 1. Precomplexes structures
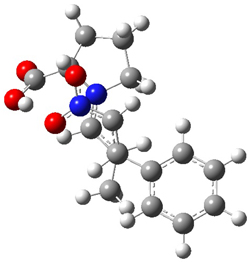
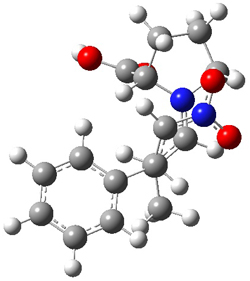
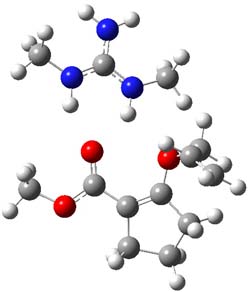
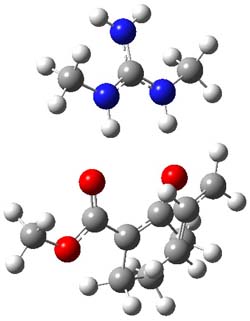
However, the lowest energy transition state takes the Grayson/Houk pathway and leads to the major isomer observed in the reaction. The Grayson/Houk TS that leads to the minor product has a barrier that is 3 kcal mol-1 higher in energy. The lowest energy TS following the Wynberg path leads to the minor product, and is 2.2 kcal mol-1 higher than the Grayson/Houk path. These transition states are shown in Figure 2. The upshot is that complex formation is not necessarily indicative of the transition state structure.
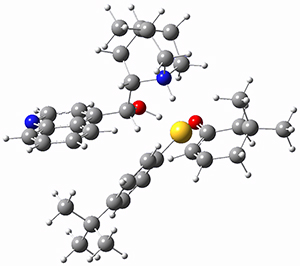
Wynberg TS (major) |
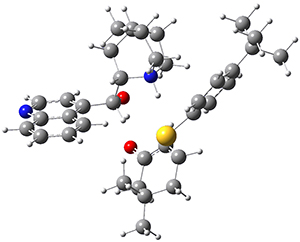
Wynberg TS (minor) |
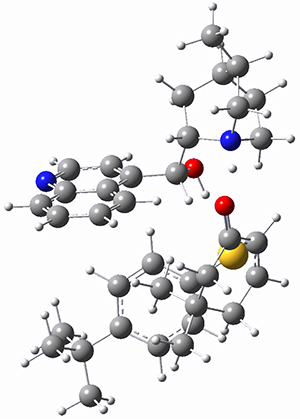
Grayson/Houk TS (major) |
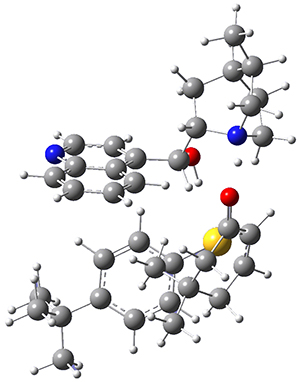
Grayson/Houk TS (minor) |
Figure 2. TS structures and relative free energies (kcal mol-1).
References
(1) Hiemstra, H.; Wynberg, H. "Addition of aromatic thiols to conjugated cycloalkenones, catalyzed by chiral .beta.-hydroxy amines. A mechanistic study of homogeneous catalytic asymmetric synthesis," J. Am. Chem. Soc. 1981, 103, 417-430, DOI: 10.1021/ja00392a029.
(2) Grayson, M. N.; Houk, K. N. "Cinchona Alkaloid-Catalyzed Asymmetric Conjugate Additions: The Bifunctional Brønsted Acid–Hydrogen Bonding Model," J. Am. Chem. Soc. 2016, 138, 1170-1173, DOI: 10.1021/jacs.5b13275.
InChIs
1: InChI=1S/C10H14S/c1-10(2,3)8-4-6-9(11)7-5-8/h4-7,11H,1-3H3
InChIKey=GNXBFFHXJDZGEK-UHFFFAOYSA-N
2: InChI=1S/C8H12O/c1-8(2)5-3-4-7(9)6-8/h3-4H,5-6H2,1-2H3
InChIKey=CDDGRARTNILYAB-UHFFFAOYSA-N
3: InChI=1S/C18H22N2O/c1-12-11-20-9-7-13(12)10-17(20)18(21)15-6-8-19-16-5-3-2-4-14(15)16/h2-6,8,12-13,17-18,21H,7,9-11H2,1H3/t12?,13?,17?,18-/m1/s1
InChIKey=ZOZLJWFJLBUKKL-NKHWWFDVSA-N
4: InChI=1S/C18H26OS/c1-17(2,3)13-6-8-15(9-7-13)20-16-10-14(19)11-18(4,5)12-16/h6-9,16H,10-12H2,1-5H3/t16-/m0/s1
InChIKey=XUTYYZOSKLYWLW-INIZCTEOSA-N