We noted in Chapter 2.1 some serious errors in the prediction of bond dissociation energies using B3LYP. For example, Gilbert examined the C-C bond dissociation energy of some simple branched alkanes.1 The mean absolute deviation (MAD) for the bond dissociation energy predicted by G3MP2 is 1.7 kcal mol-1 and 2.8 kcal mol-1 using MP2. In contrast, the MAD for the B3LYP predicted values is 13.7 kcal mol-1, with some predictions in error by more than 20 kcal mol-1. Furthermore, the size of the error increases with the size of the molecule. Consistent with this trend, Curtiss and co-workers noted a systematic underestimation of the heat of formation of linear alkanes of nearly 0.7 kcal mol-1 per bond using B3LYP.2
Further evidence disparaging the general performance of DFT methods (and B3LYP in particular) was presented in a paper by Grimme and in two back-to-back Organic Letters articles, one by Schreiner and one by Schleyer. Grimme3 noted that the relative Energy of two C8H18 isomers, octane and 2,2,3,3-tetramethylbutane are incorrectly predicted by DFT methods (Table 1). While MP2 and CSC-MP2 (spin-component-scaled MP2) correctly predict that the more branched isomer is more stable, the DFT methods predict the inverse! Grimme attributes this error to a failure of these DFT methods to adequately describe medium-range electron correlation.
|
Table 1. Energy (kcal mol-1) of 2,2,3,3-tetramethylbutane relative to octane. |
||||||||||||||||||||
|
||||||||||||||||||||
|
aNIST Webbook (http://webbook.nist.gov) bRef. 3. cUsing the cQZV3P basis set and MP2/TZV(d,p) optimized geometries. dRef. 4. eCalculated at M05-2X/6-311+G(2df,2p). |
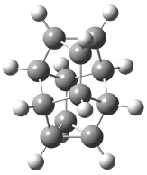
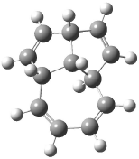
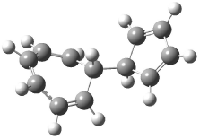
Schreiner5 also compared the energies of hydrocarbon isomers. For example, the three lowest energy isomers of C12H12 are 1-3, whose B3LYP/6-31G(d) structures are shown in Figure 1. What is disturbing is that the relative energies of these three isomers depends strongly upon the computational method (Table 2), especially since these three compounds appear to be quite ordinary hydrocarbons. CCSD(T) predicts that 2 is about 15 kcal mol-1 less stable than 1 and that 3 lies another 10 kcal mol-1 higher in energy. MP2 exaggerates the separation by a few kcal mol-1. HF predicts that 1 and 2 are degenerate. The large HF component within B3LYP leads to this DFT method’s poor performance. B3PW91 does reasonably well in reproducing the CCSD(T) results.

|
Table 2. Energies (kcal mol-1) of 2 and 3 relative to 1. |
|||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||
|
|
1 |
2 |
3 |
Figure 1. Structures of 1-3 at B3LYP/6-31G(d).
Another of Schreiner’s examples is the relative energies of the C10H10 isomers; Table 3 compares their relative experimental heats of formation with their computed energies. MP2 adequately reproduces the isomeric energy differences. B3LYP fairs quite poorly in this task. The errors seem to be most egregious for compounds with many single bonds. Schreiner recommends that while DFT-optimized geometries are reasonable, their energies are unreliable and some non-DFT method should be utilized instead.
|
Table 3. Relative C10H10 isomer energies (kcal mol-1)5 |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
aCorrelation coefficient between Rel. ΔHf and Rel. E(B3LYP). bCorrelation coefficient between Rel. ΔHf and Rel. E(MP2). |



Schleyer’s example of poor DFT performance is in the isodesmic energy of Reaction 1 evaluated for the n-alkanes.6 The energy of this reaction becomes more positive with increasing chain length, which Schleyer attributes to stabilizing 1,3-interactions between methyl or methylene groups. (Schleyer ascribes the term “protobranching” to this phenomenon.) The stabilization energy of protobranching using experimental heats of formation increases essentially linearly with the length of the chain, as seen in Figure 2.
n-CH3(CH2)mCH3 + mCH4 → (m + 1)C2H6 Reaction 1
Schleyer evaluated the protobranching energy using a variety of methods, and these energies are also plotted in Figure 2. As expected, the G3 predictions match the experimental values quite closely. However, all of the DFT methods underestimate the stabilization energy. Most concerning is the poor performance of B3LYP. All three of these papers clearly raise concerns over the continued widespread use of B3LYP as the de facto DFT method. Even the new hybrid meta-GGA functionals fail to adequately predict the protobranching phenomenon, leading Schleyer to conclude: “We hope that Check and Gilbert’s pessimistic admonition that ‘a computational chemist cannot trust a one-type DFT calculation’1 can be overcome eventually”. These papers provide a clear challenge to developers of new functionals.

Figure 2. Comparison of computed and experimental protobranching stabilization energy (as defined in Reaction 1) vs. m, the number of methylene groups of the n-alkane chain.6
Truhlar believes that one of his newly developed functionals answers the call for a reliable method. In a recent article,4 Truhlar demonstrates that the M05-2X7 functional performs very well in all three of the cases discussed here. In the case of the C8H18 isomers (Table 1), M05-2X properly predicts that 2,2,3,3-tetramethylbutane is more stable than octane, and estimates their energy difference within the error limit of the experiment. Second, M05-2X predicts the relative energies of the C12H12 isomers 1-3 within a couple of kcal mol-1 of the CCSD(T) results (see Table 2). Last, in evaluating the isodesmic energy of Reaction 1 for hexane and octane, M05-2X/6-311+G(2df,2p) predicts energies of 11.5 and 17.2 kcal mol-1 respectively. These are in excellent agreement with the experimental values of 13.1 kcal mol-1 for butane and 19.8 kcal mol-1 for octane.
Truhlar has also touted the M05-2X functional’s performance in handling noncovalent interactions.8 For example, the mean unsigned error (MUE) in the prediction of the binding energies of six hydrogen-bonded dimers is 0.20 kcal mol-1. This error is comparable to that from G3 and much better than CCSD(T). With the M05-2X functional already implemented within NWChem and soon to be released within Gaussian and Jaguar, it is likely that M05-2X may supplant B3LYP as the new de facto functional in standard computational chemical practice.
Schleyer has now examined the bond separation energies of 72 simple organic molecules computed using a variety of functionals,9 including the workhorse B3LYP and Truhlar’s new M05-2X. Bond separation energies are defined by reactions of each compound, such as three shown below:

The new M05-2X functional performed the best, with a mean absolute deviation (MAD) from the experimental energy of only 2.13 kcal mol-1. B3LYP performed much worse, with a MAD of 3.96 kcal mol-1. As noted before, B3LYP energies become worse with increasing size of the molecules, but this problem is not observed for the other functionals examined (including PW91, PBE, and mPW1PW91, among others). So while M05-2X overall appears to solve many of the problems noted with common functionals, it too has some notable failures. In particular, the error is the bond separation energies of 4, 5, and 6 is -8.8, -6.8, and -6.0 kcal mol-1, respectively.
References
(1) Check, C. E.; Gilbert, T. M., “Progressive Systematic Underestimation of Reaction Energies by the B3LYP Model as the Number of C-C Bonds Increases: Why Organic Chemists Should Use Multiple DFT Models for Calculations Involving Polycarbon Hydrocarbons,” J. Org. Chem. 2005, 70, 9828-9834, DOI: 10.1021/jo051545k.
(2) Redfern, P. C.; Zapol, P.; Curtiss, L. A.; Raghavachari, K., “Assessment of Gaussian-3 and Density Functional Theories for Enthalpies of Formation of C1-C16 Alkanes,” J. Phys. Chem. A 2000, 104, 5850-5854, DOI: 10.1021/jp994429s.
(3) Grimme, S., “Seemingly Simple Stereoelectronic Effects in Alkane Isomers and the Implications for Kohn-Sham Density Functional Theory,” Angew. Chem. Int. Ed. 2006, 45, 4460-4464, DOI: 10.1002/anie.200600448
(4) Zhao, Y.; Truhlar, D. G., “A Density Functional That Accounts for Medium-Range Correlation Energies in Organic Chemistry,” Org. Lett. 2006, 8, 5753-5755, DOI: 10.1021/ol062318n
(5) Schreiner, P. R.; Fokin, A. A.; Pascal, R. A.; deMeijere, A., “Many Density Functional Theory Approaches Fail To Give Reliable Large Hydrocarbon Isomer Energy Differences,” Org. Lett. 2006, 8, 3635-3638, DOI: 10.1021/ol0610486
(6) Wodrich, M. D.; Corminboeuf, C.; Schleyer, P. v. R., “Systematic Errors in Computed Alkane Energies Using B3LYP and Other Popular DFT Functionals,” Org. Lett. 2006, 8, 3631-3634, DOI: 10.1021/ol061016i
(7) Zhao, Y.; Schultz, N. E.; Truhlar, D. G., “Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions,” J. Chem. Theory Comput. 2006, 2, 364-382, DOI: 10.1021/ct0502763.
(8) Zhao, Y.; Truhlar, D. G., “Assessment of Model Chemistries for Noncovalent Interactions,” J. Chem. Theory Comput. 2006, 2, 1009-1018, DOI: 10.1021/ct060044j.
(9) Wodrich, M. D.; Corminboeuf, C.; Schreiner, P. R.; Fokin, A. A.; Schleyer, P. v. R., “How Accurate Are DFT Treatments of Organic Energies?,” Org. Lett., 2007, 9, 1851-1854, DOI: 10.1021/ol070354w.
InChI
1: InChI=1/C11H10/c1-2-5-7-3(1)4(1)8-6(2)10-9(5)11(7,8)10/h1-10H
2: InChI=1/C12H12/c1-2-4-10-6-8-11-7-5-9(3-1)12(10)11/h1-12H
3: InChI=1/C12H12/c1-2-4-8-11(7-3-1)12-9-5-6-10-12/h1-12H
4: InChI=1/C6H6/c1-4-5(2)6(4)3/h1-3H2
5: InChI=1/C8H10/c1-3-7-5-6-8(7)4-2/h3-4H,1-2,5-6H2
6: InChI=1/C10H10/c1-2-8-5-6-9-4-3-7(1)10(8)9/h1-10H

Gabor I. Csonka responded on 21 Dec 2007 at 4:51 am #
I would like to note that CCSD(T)/cc-pVDZ energies in Table 2 for 2 and 3 are far from being converged. Our study shows that these energies are about 12.2 and 20.5 kcal/mol (CBS extrapolated CCSD(T) results), respectively. The details will be published soon. MP2 relative energies are in serious error for these relative energies, so I question the use of MP2 as a reference for such energy differences.
Computational Organic Chemistry » SN2 and E2 DFT benchmark responded on 22 Jul 2008 at 8:45 am #
[…] There is a lot more data in this paper, along with a summary of the mean absolute errors in the overall and central barriers that mimics the data I show in Table 1. The trends are pretty clear. Generalized gradient approximation (GGA) functions – like BLYP, PW91, and PBE – dramatically underestimate the barriers. The hybrid functionals perform much better. The recently maligned B3LYP functional gets the correct trend and provides reasonable estimates of the barriers. Truhlar’s MO5-2X and MO6-2X functionals do very well in matching up the barrier heights along with getting the correct trends in the relative barriers. Simply looking for the functional with the lowest absolute error is not sufficient; BHandH and MO6-L have small errors but give a wrong trend in barriers, predicting that the SN2 reaction is preferred over the E2 for the fluoride reaction. […]
Computational Organic Chemistry » Origin of DFT failure responded on 19 May 2009 at 8:15 am #
[…] some seemingly straightforward reactions (as discussed in these previous blog posts: A, B, C, D, E, F) has become a bit clearer. Brittain and co-workers have identified the culprit.1 They examined […]
Gabor I. Csonka responded on 11 Aug 2010 at 12:53 pm #
See our work about protobranching (a new version of the dispersion correction is also out):
Unified Inter- and Intramolecular Dispersion Correction Formula for Generalized Gradient Approximation Density Functional Theory: J. Chem. Theory Comput., 2009, 5 (11), pp 2950–2958, DOI: 10.1021/ct9002509
Steven Bachrach responded on 11 Aug 2010 at 12:57 pm #
Gabor – sorry I missed this article!