What is the relationship between a ground state and the first excited triplet (or first excited singlet) state regarding aromaticity? Baird1 argued that there is a reversal, meaning that a ground state aromatic compound is antiaromatic in its lowest triplet state, and vice versa. It is suggested that the same reversal is also true for the second singlet (excited singlet) state.
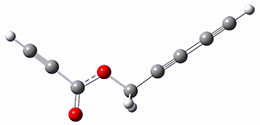
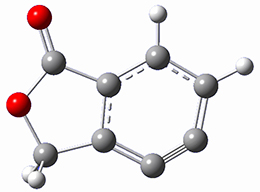
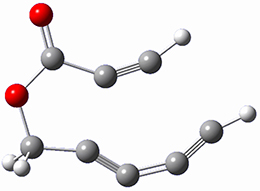
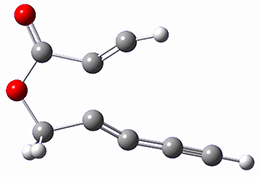
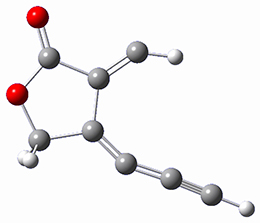
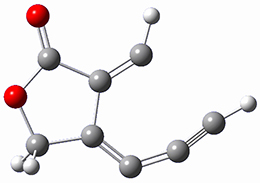
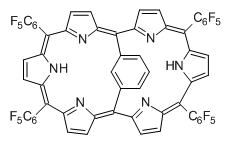
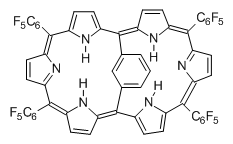
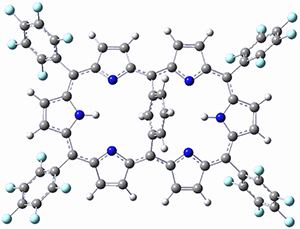
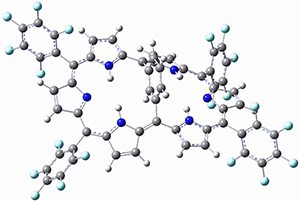
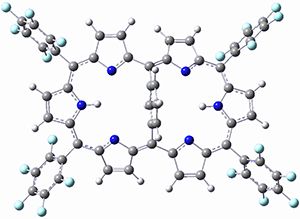
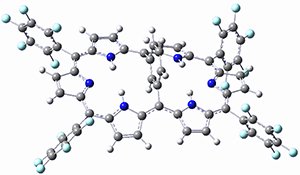
Osuka, Sim and coworkers have examined the geometrically constrained hexphyrins 1 and 2.2 1 has 26 electrons in the annulene system and thus should be aromatic in the ground state, while 2, with 28 electrons in its annulene system should be antiaromatic. The ground state and lowest triplet structures, optimized at B3LYP/6-31G(d,p), of each of them are shown in Figure 1.
|
|
|
11 |
12 |
31 |
32 |
Figure 1. B3LYP/6-31G(d,p) optimized geometries of 1 and 2.
NICS computations where made in the centers of each of the two rings formed by the large macrocycle and the bridging phenyl group (sort of in the centers of the two lenses of the eyeglass). The NICS values for 1 are about -15ppm, indicative of aromatic character, while they are about +15ppm for 2, indicative of antiaromatic character. However, for the triplet states, the NICS values change sign, showing the aromatic character reversal between the ground and excited triplet state. The aromatic states are also closer to planarity than the antiaromatic states (which can be seen by clicking on the images in Figure 1, which will launch the JMol applet so that you can rotate the molecular images).
They also performed some spectroscopic studies that support the notion of aromatic character reversal in the excited singlet state.
References
(1) Baird, N. C. "Quantum organic photochemistry. II. Resonance and aromaticity in the lowest 3ππ* state of cyclic hydrocarbons," J. Am. Chem. Soc. 1972, 94, 4941-4948, DOI: 10.1021/ja00769a025.
(2) Sung, Y. M.; Oh, J.; Kim, W.; Mori, H.; Osuka, A.; Kim, D. quot;Switching between Aromatic and Antiaromatic 1,3-Phenylene-Strapped [26]- and [28]Hexaphyrins upon Passage to the Singlet Excited State," J. Am. Chem. Soc. 2015, 137, 11856-11859, DOI: 10.1021/jacs.5b04047.
InChIs
1: InChI=1S/C60H18F20N6/c61-41-37(42(62)50(70)57(77)49(41)69)33-23-8-4-19(81-23)31-17-2-1-3-18(16-17)32(21-6-10-25(83-21)35(29-14-12-27(33)85-29)39-45(65)53(73)59(79)54(74)46(39)66)22-7-11-26(84-22)36(40-47(67)55(75)60(80)56(76)48(40)68)30-15-13-28(86-30)34(24-9-5-20(31)82-24)38-43(63)51(71)58(78)52(72)44(38)64/h1-16,85-86H/b31-19+,31-20+,32-21+,32-22+,33-23+,33-27+,34-24+,34-28+,35-25+,35-29+,36-26+,36-30+
InChIKey=TUOMWLSCXXODFY-CQGNQUHXSA-N
2: InChI=1S/C60H20F20N6/c61-41-37(42(62)50(70)57(77)49(41)69)33-23-8-4-19(81-23)31-17-2-1-3-18(16-17)32(21-6-10-25(83-21)35(29-14-12-27(33)85-29)39-45(65)53(73)59(79)54(74)46(39)66)22-7-11-26(84-22)36(40-47(67)55(75)60(80)56(76)48(40)68)30-15-13-28(86-30)34(24-9-5-20(31)82-24)38-43(63)51(71)58(78)52(72)44(38)64/h1-16,81-84H/b31-19+,31-20+,32-21+,32-22+,33-23+,33-27+,34-24+,34-28+,35-25+,35-29+,36-26+,36-30+
InChIKey=KTIAGNMFTAGKFJ-CQGNQUHXSA-N