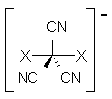
Shaik, Wu and Hiberty have proposed a third bond type, and they have a nice review article in Nature Chemistry.1 Along with the long-standing concepts of the covalent bond and the ionic bond, they add a third category: the charge-shift bond.
The valence bond wavefunction for the diatomic A-B is written as
Ψ(VB) = c1φcov(A-B) + c2φion(A+B–) + c3φion(A–X+)
Typically one of these terms dominates and we call the bond covalent if c1 is the largest coefficient or ionic if either c2 or c3 is the largest term. The bond dissociation energy (De) is the difference in energy of the total VB wavefunction (above) and the energy of the separate radicals A. and B.. One can determine the energy due to just a single component of the total VB wavefunction. One might expect that for a covalent bond, the bond dissociation energy derived from just the c1φcov(A-B) term would be close to De. For many covalent bonds this is true. However, Shaik and co-authors show a number of bonds where this is not true. For example, in the F-F bond, the covalent term is destabilizing. Rather, it is the resonance energy due to the mixing of the 3 VB terms that leads to bond formation. Shaik, Wu and Hiberty call this the “charge-shift bond”. They describe a number of examples of typically understood homonuclear and heteronuclear covalent bonds that are in fact charge-shift bonds, and an example of an ionic bond that really is charge-shift.
They argue that the charge-shift bond manifests as a consequence of the virial theorem. When an atom participates in a bond, its size gets smaller and this results in an increase in its kinetic energy. If the atom gets very small, then a substantial resultant change in the potential energy must occur, and this is the charge-shift bond. This also occurs in bonds involving atoms with many lone pairs; the lone-pair bond-weakening effect also causes a rise in kinetic energy that must be offset.
The authors speculate that many more examples of the charge-shift bond are waiting to be uncovered. It will be interesting if this concept catches hold and how quickly it will incorporated into general chemistry textbooks.
References
(1) Shaik, S.; Danovich, D.; Wu, W.; Hiberty, P. C., "Charge-shift bonding and its manifestations in chemistry," Nature Chem., 2009, 1, 443-449, DOI: 10.1038/nchem.327