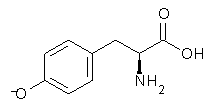
Organocatalysis affected by proline is an extremely active research area, and computational chemists have made considerable contributions (see Chapter 5.3 of my book – and this is expanded on in the 2nd edition which should be out in just a few months). Most importantly, the Houk-List model1 for the catalysis was largely developed on the basis of computations.
Recent experiments have indicated cocatalysts that can hydrogen bond to proline may increase the catalytic effect, including the enantioselectivity. Xue and co-workers have examined a series of potential cocatalysts for their ability to enhance the acidity of proline.2 This is important in that a proton transfer is a component to the key step of the Houk-List model.
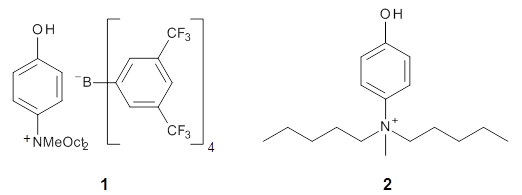
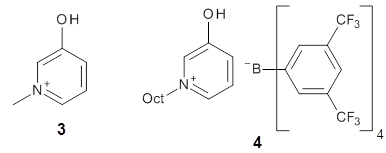
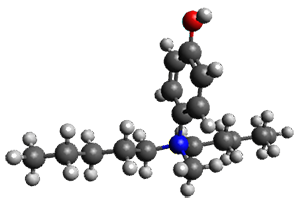
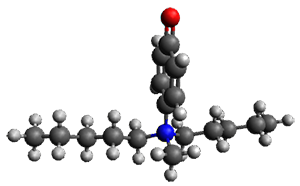
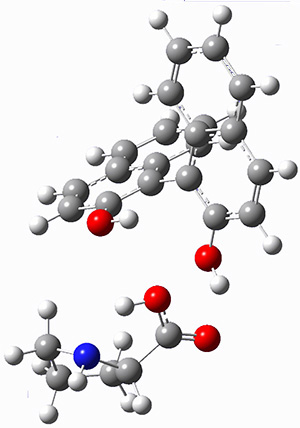
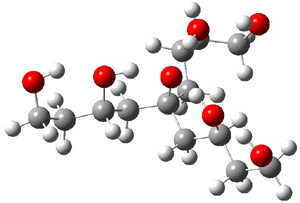
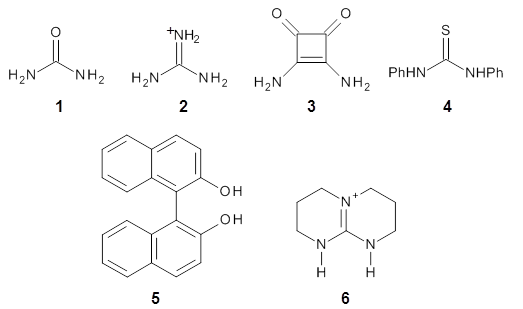
The cocatalysts examined included such compounds as 1–6. The deprotonation energy of proline with the associated cocatalysts was compared with that of proline itself. The energies were computed at M06-2x/6-311++G(2df,2p)//B3LYP/6-31+G(d) with the SMD treatment of five solvents. The structure of 5 with proline is shown in Figure 1.
|
5 with proline
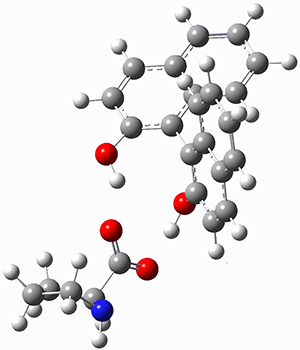
|
5 with proline conjugate base
|
Figure 1. M06-2x/6-311++G(2df,2p)//B3LYP/6-31+G(d) optimized structure of 5 with proline and its conjugate base.
The effect of the cocatalysts is striking. In the gas phase, these additives decrease the pKa of proline by 15 – 70 pKa units, with 2 showing the largest effect. In solvent, the effect of the cocatalyst is attenuated, especially in more polar solvents, but still a considerable decrease in the pKa is seen (as much as a 12 pKa unit increase in acidity). Further exploration of potential cocatalysts seems fully warranted.
References
(1) Allemann, C.; Gordillo, R.; Clemente, F. R.; Cheong, P. H.-Y.; Houk, K. N. "Theory of Asymmetric Organocatalysis of Aldol and Related Reactions: Rationalizations and Predictions," Acc. Chem. Res. 2004, 37, 558-569, DOI: 10.1021/ar0300524.
(2) Xue, X.-S.; Yang, C.; Li, X.; Cheng, J.-P. "Computational Study on the pKa Shifts in Proline Induced by Hydrogen-Bond-Donating Cocatalysts," J. Org. Chem. 2014, 79, 1166–1173, DOI: 10.1021/jo402605n.
InChIs
1: InChI=1S/CH4N2O/c2-1(3)4/h(H4,2,3,4)
InChIKey=XSQUKJJJFZCRTK-UHFFFAOYSA-N
2: InChI=1S/CH5N3/c2-1(3)4/h(H5,2,3,4)/p+1
InCiKey=ZRALSGWEFCBTJO-UHFFFAOYSA-O
3: InChI=1S/C4H4N2O2/c5-1-2(6)4(8)3(1)7/h5-6H2
InChIKey=WUACDRFRFTWMHE-UHFFFAOYSA-N
4: InChI=1S/C13H12N2S/c16-13(14-11-7-3-1-4-8-11)15-12-9-5-2-6-10-12/h1-10H,(H2,14,15,16)
InChIKey=FCSHMCFRCYZTRQ-UHFFFAOYSA-N
5: InChI=1S/C20H14O2/c21-17-11-9-13-5-1-3-7-15(13)19(17)20-16-8-4-2-6-14(16)10-12-18(20)22/h1-12,21-22H
InChIKey=PPTXVXKCQZKFBN-UHFFFAOYSA-N
6: InChI=1S/C7H13N3/c1-3-8-7-9-4-2-6-10(7)5-1/h1-6H2,(H,8,9)/p+1
InChIKey=FVKFHMNJTHKMRX-UHFFFAOYSA-O
Proline: InChI=1S/C5H9NO2/c7-5(8)4-2-1-3-6-4/h4,6H,1-3H2,(H,7,8)/t4-/m0/s1
InChIKey= ONIBWKKTOPOVIA-BYPYZUCNSA-N