I think most organic chemists hold dear to their hearts the notion that selectivity is due to crossing over different transition states. Readers of my book and this blog know of the many examples where this notion simply is not true (see here). This post discusses yet another example taking place in a seemingly simple reaction.
Singleton has examined the nucleophilic substitution reaction of 1 with sodium tolylsulfide.1 The mono substitution gives potentially two different stereoproducts 2 and 3. The experimental ratio of these products 2:3 is 81:19. (Note that things are a bit more complicated because disubstitution can also occur, but this has been factored into the product ratio.)
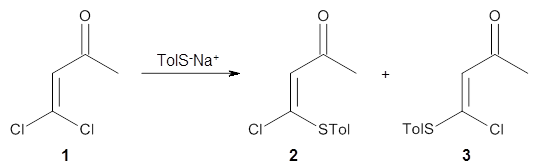
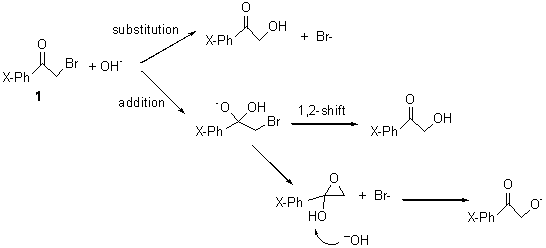
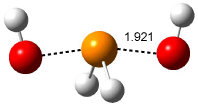
Based on previous literature, this reaction is likely to proceed in a concerted fashion, and so one might anticipate running computations to locate a transition state leading to 2 and a transition state leading to 3. In fact, Singleton finds six different TSs (the lowest energy TS 4 is shown in Figure 1), all within 2 kcal mol-1 of each other at PCM(ethanol)/B3LYP/6-31+G**. However, the intrinsic reaction coordinate going forward from each of these six TSs leads solely to 2; no TS could be located that connects to 3! (Computations were also performed at PCM(ethanol)/M06-2x/6-31+G** which give very similar results.) Classical transition state theory would lead
one to conclude that only 2 should be formed, inconsistent with experiment.
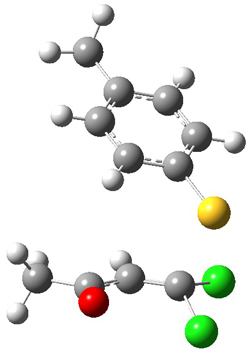 4 |
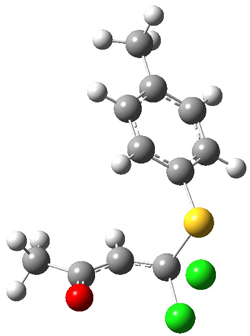 5 |
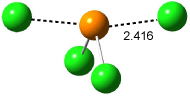
Figure 1. PCM/B3LYP/6-31+G** optimized structures of TSs 4 and 5.
Furthermore, no intermediate could be located. This is consistent with a concerted mechanism. A second transition state was located which interconverts 2 and 3 with the involvement of a chloride – a sort of addition/rotation/elimination process. This TS 5 is also shown in Figure 1.
A direct dynamics study was performed, and 197 trajectories were computed. Of these, 185 trajectories went to product: 156 to 2 and 29 to 3, for a ratio of 84:16 – in amazing agreement with experiment! The product selectivity is due entirely to dynamic effects. In fact, it is one vibrational mode that dictates the product distribution. Essentially, the nature of the rotation about the C=C bond differentiates the eventual route, with a clockwise rotation leading always to 2 and a counterclockwise rotation leading about a third of the time to 3.
References
(1) Bogle, X. S.; Singleton, D. A. "Dynamic Origin of the Stereoselectivity of a Nucleophilic Substitution Reaction," Org. Lett., 2012, 14, 2528-2531, DOI: 10.1021/ol300817a.
InChIs
1: InChI=1S/C4H4Cl2O/c1-3(7)2-4(5)6/h2H,1H3
InChIKey=NXDUHPYJFYSBCT-UHFFFAOYSA-N
2: InChI=1S/C11H11ClOS/c1-8-3-5-10(6-4-8)14-11(12)7-9(2)13/h3-7H,1-2H3/b11-7-
InChIKey=NCXXSKTZGJETLW-XFFZJAGNSA-N
3: InChI=1S/C11H11ClOS/c1-8-3-5-10(6-4-8)14-11(12)7-9(2)13/h3-7H,1-2H3/b11-7+
InChIKey=NCXXSKTZGJETLW-YRNVUSSQSA-N