The search for ever more intriguing aromatic/antiaromatic species continues on – Haley has recently prepared the TIPS-protected indeno[1,2-b]fluorene 1. 1 The crystal structure was determined and analogue with the tri-iso-propylsilyl groups replaced with hydrogens (2) has been computed at B3LYP/6-31G(d,p). This optimized structure is shown in Figure 1. The core system has 20 π-electrons – suggesting perhaps an antiaromatic system.
|
|
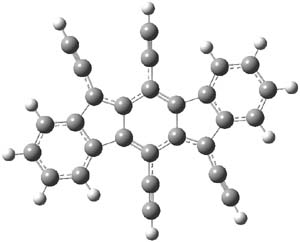 2 |
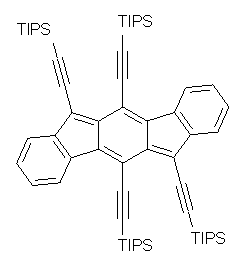
Figure 1. B3LYP/6-31G(d,p) optimized geometry of 2.
The x-ray structure and computed structure are in close agreement in terms of distances. The terminal phenyl rings exhibit very little alternation. The C1-C2 and C2-C3 distances are long (1.444 and 1.457 Å, respectively) while the C1-C3A and C2-C4 distances are short (1.379 and 1.396 Å, respectively.) This suggests a para-xylylene type structure for the central six-member ring. The NICS values of the terminal 6-member ring, the 5-member ring and the central 6-member ring are computed to be -7.12 (a reasonable phenyl value), 1.84, and 0.02. So the middle three rings possess no aromatic or antiaromatic character. Haley describes this structure as “a fully conjugated 20-π-electron hydrocarbon with fused s-trans 1,3-diene linkages across the top and bottom portions of the carbon skeleton”.
References
(1) Chase, D. T.; Rose, B. D.; McClintock, S. P.; Zakharov, L. N.; Haley, M. M., "Indeno[1,2-b]fluorenes: Fully Conjugated Antiaromatic Analogues of Acenes," Angew. Chem. Int. Ed., 2011, 50, 1127-1130, DOI: 10.1002/anie.201006312
InChIs
1: InChI=1/C64H92Si4/c1-41(2)65(42(3)4,43(5)6)37-33-57-53-29-25-27-31-55(53)61-60(36-40-68(50(19)20,51(21)22)52(23)24)64-58(34-38-66(44(7)8,45(9)10)46(11)12)54-30-26-28-32-56(54)62(64)59(63(57)61)35-39-67(47(13)14,48(15)16)49(17)18/h25-32,41-52H,1-24H3
InChIKey=JIXLLIXVMPJPEE-UHFFFAOYAI
2: InChI=1/C28H12/c1-5-17-21-13-9-11-15-23(21)27-20(8-4)26-18(6-2)22-14-10-12-16-24(22)28(26)19(7-3)25(17)27/h1-4,9-16H
InChIKey=DOQPHDKEGVECBF-UHFFFAOYAY
